 |
History
A 52-year-old white female presented to the office with a chief complaint of blurry vision in both eyes at distance and near. She reported that the right eye had been blurry for months and the left eye became blurred one month prior to this appointment. She reported no use of ocular medications; her systemic medications consisted of bisacodyl for constipation. She reported no known allergies to medications or anything else.
Diagnostic Data
Her best-corrected entering visual acuities were 20/80 OD and 20/150 OS. Her pupils were round, equal in size and reactive to light without an afferent pupillary defect. Extraocular muscles were full and confrontation visual fields were full to finger counting with a central blur OU when performing the “facial” Amsler. Refraction was completed with no improvement at either distance or near. Biomicroscopy was remarkable only for trace nuclear sclerosis in both eyes.
Her intraocular pressures using Goldmann applanation tonometry measured 13mm Hg OD and 12mm Hg.
Your Diagnosis
Does the case presented require any additional tests, history or information? What steps would you take to manage this patient? Based on the information provided, what would be your diagnosis? What is the most likely prognosis?
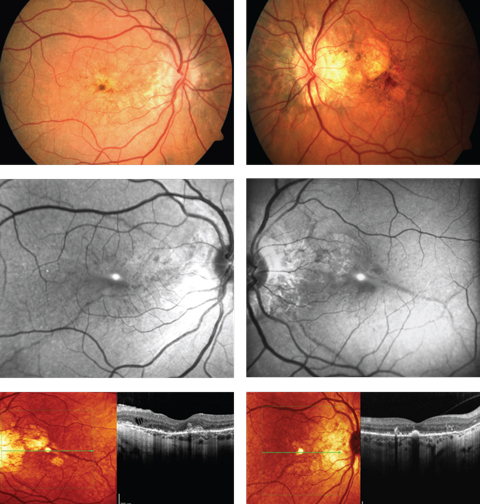 |
| A 52-year-old woman complained of progressive blurriness in both eyes. What can these images and her history combined tell you about her possible diagnosis? Click images to enlarge. |
Diagnosis and Discussion
The diagnosis in this case is bilateral choroidal neovascularization (CNV) secondary to angioid streaks (AS). The patient was referred to retinology to rule out vascular endothelial growth factor inhibitor injections vs. laser photoablation consistent with macular photocoagulation ptudy or photodynamic therapy. While angioid streaks are common in patients with moderate to high myopia and can occur idiopathically, they are also known to occur in patients with systemic disease (sickle cell anemia, pseudoxanthoma elasticum, Paget’s disease of the bone, Marfan’s syndrome and Ehlers-Danlos disease). A referral to the internist was made with correspondence and photographs explaining the common systemic diseases that produce them suggesting an appropriate work up be completed. Systemic testing uncovered pseudoxanthoma elasticum (PXE), also known as Grönblad–Strandberg syndrome.
PXE or Grönblad–Strandberg syndrome is an autosomal recessive genetic disorder characterized by degeneration and mineralization of the body’s elastin fibers.1-8 Elastin is a protein found throughout the body including the skin, eyes, and blood vessels. Researchers estimate the prevalence of PXE at 1 in 25,000 to 100,000.1 They also estimate that women are twice as likely to inherit PXE as men.1 The reason for the female predilection is not entirely understood.1-8 The diagnosis of PXE is made by skin biopsy or genetic testing. Typically, stains such as Von Kossa or Verhoeff’s stains are used to evaluate samples for calcium or elastin. A defect in the ABCC6 gene on chromosome 16p13.1 has been associated its inheritance.2 The ABCC6 gene is an organic anion transporter protein with defects increasing the risk of expression of PXE.3
Those affected by PXE can present with a myriad of systemic findings. PXE can affect a wide range of organs, primarily targeting the skin, cardiovascular system, and eyes.1-15 Gastrointestinal hemorrhage is another reported complication.3 Early dermatological findings include yellowish, raised lesions, typically observed around the neck.3 These lesions are often described as the “plucked chicken” appearance.3 Multiple skin folds, typically located around the neck or armpit region are also common.3 Due to excess arteriolar calcification of small to medium sized arteries, individuals will sometimes suffer with complications related to the cardiovascular system. Cardiovascular complications include, but are not limited to, angina pectoris, hypertension, mitral-valve prolapse and restrictive cardiomyopathy.3 The excessive calcification of arteries increases the risk of myocardial infarction in these patients.3
Ocular findings include: AS with secondary CNV, the peau d’orange appearance in the posterior pole of the fundus, drusen of the optic nerve head and comet tail lesions (chorioretinal atrophy, salmon spots).4 Additionally, more than 70% of eyes in patients with PXE showed degenerative changes of the retinal pigment epithelium (RPE).5 The peau d’orange appearance of the fundus or leopard skin spotting variation is observable as mottling of the retina temporal to the macula. This phenomenon appears to precede the formation of AS and can be found in patients as young as 10.14,15 Spontaneous degradation of the RPE and Bruch’s membrane complex can lead to geographic atrophy with or without the presence of CNV. The mechanism of this atrophy is not well understood.14 Finally, those with PXE are prone to the formation of optic nerve head drusen. It is estimated that 0.3% of individuals in the general population have optic nerve head drusen compared to roughly 20% of PXE patients.15
AS are the most renowned ocular finding in PXE, often appearing between the ages of 15 to 25 years.6 They are referred to as “angioid” due to the resemblance they share with blood vessels. Histologically, AS are the result of 50-500um of thickening, or more specifically, generalized heavy calcification of elastin fibers in Bruch’s membrane. These fibers are colored reddish brown and emanate from the optic nerve creating the appearance of streaks.2,4 As a consequence, affected regions of the retina become structurally compromised as localized atrophy of the RPE and choriocapillaris evolves. This is also the impetus for the creation of choroidal neovascular membrane (CNV) formation.4 AS can be differentiated funduscopically from blood vessels by their depth (AS lie deeper than the retinal blood vessels and will thus cross underneath them).
The prevalence of AS in individuals with PXE approaches 100%.7,8 It is estimated that the development of CNV in patients who have AS varies between 72% to 86%.4 Since Bruch’s membrane becomes brittle from mineralization, individuals are more prone to retinal hemorrhage and CNV from even the most minor sources of trauma.9 By comparison studies have shown that the incidence of CNV after trauma is around 15%.10
Although many systemic conditions are associated with AS, pseudoxanthoma elasticum, Ehlers-Danlos syndrome, Paget’s disease of the bone and the sickle-cell hemoglobinopathies are the notable differentials.2 They may also occur idiopathically and in association with myopia.
Ehlers-Danlos syndrome is a heritable group of connective tissue disorders which affect the collagen integrity of bone, skin, blood vessels, and other organs.11,12 Affected individuals have skin hyperextensibility and fragility as well as fragile bones.11 Paget’s disease is a genetic, metabolic bone disease in which there is chronic bone resorption and regrowth resulting in a structurally weakened and more vascular skeleton.12 Affected individuals can present with hearing loss and deformities of the skull and weight bearing bones.12
Sickle cell hemoglobinopathes affect red blood cells causing them to become crescent shaped and oxygen deprived. AS in these patients are hypothesized to be correlated with calcium deposition at the level of Bruch’s membrane.2 Other notable ocular findings include salmon patch hemorrhages, vascular occlusions and intra-retinal “sea fan” appearing neovascularization.13
The mnemonic PEPSI (Paget’s, Ehlers, Pseudoxanthoma, Sickle Cell and Idiopathic) is an effective tool for quickly recalling the major etiologies associated with AS.
Fundus imaging is useful in highlighting the different pathological retinal processes in these cases and includes fundus autofluorescence (FAF), cross-sectional and “en face” optical coherence tomography (OCT), and optical coherence tomography angiography (OCT-A).1-17 FAF permits the examiner the ability to examine the retinal pigment epithelium (RPE) and photoreceptors for damage.2 Specifically, FAF highlights over-production and accumulation of pathological fluorophores in the RPE, such as lipofuscin; increased amounts can be attributable to certain ocular diseases.16 Fluorophores emit specific wavelength light that can be captured and quantified in response to exposure to a reference wavelength of light. Positive FAF images demonstrate hyperfluorescence and hypofluorescence. Hyperfluorescence results from increased production of lipofuscin, retinal drusen or defects in the photoreceptors and RPE causing a window type defect.16,17 Lipofuscin is a metabolic waste product produced by the RPE cells from breakdown of photoreceptor outer segments. This is normally a product of the retina, but accumulation of this by-product can be a sign of damage to the deep retina. On FAF AS appear as attenuated/hypofluorescent streaks due to injury to RPE cells and subsequent loss of pigment granules.2 Cross-sectional and en face OCT in patients with PXE shows deep retinal and choroidal changes.18,19 These changes include: Hyper-reflective lesions in the subretinal space resembling subretinal drusenoid deposits, breaks in Bruch’s membrane with preservation of overlying RPE, pockets of subretinal fluid not associated with CNV, outer retinal tubulation and absence of visualization of the external limiting membrane or photoreceptor integrity line.18 En face OCT has been specifically shown to locate AS as a structural defect in Bruch’s membrane, as well as to determine the spatial relationship between AS and forming CNV.19
OCT-A is a non-invasive imaging technique that demonstrates flow characteristics of the retinal and choroidal vasculature network with greater precision than dye angiography.8 OCT-A has been proven to delineate CNVs closely correlated to the site of AS and adds an additional tool to monitor these patients.8
Ocular treatment of AS involve the use of anti-VEGF agents for treating potential CNV. There are no known contraindications for the treatment of CNV in patients with AS or PXE.20-23 Studies have shown that ranibizumab, bevacizumab, and aflibercept are all safe and efficacious in the treatment of CNV and for use in the reduction of sub-retinal fluid in patients with PXE and AS.20-23 Patients should be aware that multiple treatments are often necessary due to AS remaining throughout their lifetime. Treatment will be useful in minimizing reoccurrences in some patients.24
Our patient underwent multiple intravitreal ranibizumab injections bilaterally resulting in presently quiescent CNVs. After vision rehabilitation this patient’s BCVAs are 20/40 OD and 20/100 OS with improved quality of life.
Dr. Gurwood thanks Brian Knight OD and Andrew Rixon OD for contributing this case.
1. Chassaing N, Martin L, Calvas P, et al. Pseudoxanthoma elastic ABCC6 mutations. J Med Genet. 2005;42(12):881–892. |
