
A 69-year-old white male presented to the Spokane VA Medical Center for the first time. His chief concern was stinging, burning and grittiness in both eyes that had been bothersome for a few months. His last eye exam was five years ago, which is when he was last prescribed a pair of spectacles.
The patient was also concerned about any visual problems related to his diagnosis of Waldenstrms macroglobulinemia (WM), a type of non-Hodgkins lymphoma that affects the IgM antibodies.
The patients ocular history was remarkable only for his being a hyperopic astigmat, as well as presbyopic.
His medical history was significant for hypertension, benign prostate hypertrophy, gastroesophageal reflux disease (GERD), bilateral hearing loss and anemia.
In 1997, the patient was diagnosed with Waldenstrms macroglobulinemia via a routine blood test at another hospital. He was treated with Leukeran (chlorambucil, GlaxoSmithKline) and prednisone for six months.
To date, the patient had been largely asymptomatic, with the exception of being anemic, and was currently untreated.
Diagnostic Data
His best-corrected visual acuity was 20/20 in both eyes. Slit lamp exam revealed a low lacrimal lake with a tear break-up time of less than four seconds O.U., and 1+ nuclear sclerotic cataracts in both eyes.
Intraocular pressures were 15mm Hg O.D. and 17mm Hg O.S. using Goldmann applanation tonometry at 2:45 p.m. Dilated fundus exam showed a cup-to-disc ratio of 0.1/0.1 with distinct margins in both eyes, as well as flat maculae.
The patient had scattered intraretinal hemorrhages in the mid-periphery of both eyes (see figures 1 and 2).
Diagnosis
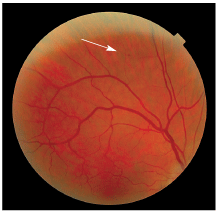
1. The patients right eye at his first visit. Note the superior dot-blot hemorrhage.
The diagnosis was retinopathy secondary to Waldenstrms macroglobulinemia, dry eye syndrome, mild senile cataracts and presbyopia.
Treatment and Follow-Up
We dispensed a new spectacle prescription and artificial tears to be used q.i.d., and we completed fundus photography. Also, we asked the patient to return in six months for a dilated fundus exam.
At the follow-up exam, the patients retinopathy had slightly worsened in both eyes, but the patient was still asymptomatic both physically and visually. Despite our best education, the patient refused fundus photography at this date, due to his concerns about the long-lasting after-images that he perceived after the previous photos were taken.
He was instructed to again return in six months for a follow-up dilated fundus exam.
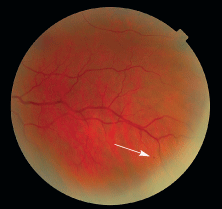
2. The patients left eye at his first visit. There is an inferior dot-blot hemorrhage.
In 1944, Swedish physician Jan Waldenstrm first described a condition that would later bear his name, where patients were suffering with anemia and epitaxis (mucosal bleeding of gums and nose).1,2 Upon physical examination, the patients also demonstrated hepatomegaly, splenomegaly and lymphadenopathy. When patients blood was examined, it was found that the presence of a then-unknown protein was causing thickening of the serum. This unknown protein would later be named immunoglobulin M (IgM).1,2
Waldenstrms macroglobulinemia is a rare, indolent form of non-Hodgkins lymphoma.3,4 About 1,500 people are diagnosed with WM every year, and most are over the age of 65, male and white.2,5 The cause of WM is still unknown, but it is characterized by the overproduction of the immunoglobulin M antibody.6 The source of the excessive IgM production is due to abnormal plasma cells that invade the bone marrow, lymph nodes and spleen (see Plasma Cell Production).2 In a handful of cases, WM has caused congestive heart failure, renal insufficiency, coma, seizures and death.7
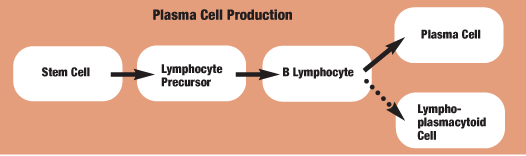
A disruption in the normal process of healthy plasma cell production results in the overproduction of IgM antibodies and Waldenstrms macroglobulinemia.2
The IgM antibody is the largest (970 kilodaltons) in the body, and approximately 80% of the molecules are found in circulation.2,3,8 Because it is the largest antibody, it is often referred to as the macroglobulin, which leads to the name Waldenstrms macroglobulinemia.2
Because of IgMs large size, high quantity, self-agglutinating properties and electrostatic charges to red blood cells, people with WM can sometimes develop hyperviscosity syndrome.8 In this syndrome, highly viscous blood causes more resistance and damages the endothelial cells that line blood vessels and causes them to leak. It interferes with blood flow through small blood vessels first, followed by larger vessels. Hyperviscosity of the blood has the potential to affect all the vital organs of the body.4,6 Hyperviscosity syndrome is commonly found in patients with WM, but also in patients with leukemia, polycythemia vera and multiple myeloma.
Positive diagnosis of Waldenstrms macroglobulinemia requires blood tests to measure total protein levels and serum viscosity, and a bone marrow biopsy is needed to confirm the diagnosis.9 Identification of WM occurs as a result of three findings:9
An immunoglobulin M concentration greater than 3,000mg/dL. (Normal range is 43mg/dl to 279mg/dL.)
Malignant lymphoplasmacytic cells found in the bone marrow.
Anemia, leukopenia or clinical impairment.8,9
Our patient developed all three characteristics: an IgM level of approximately 5,900mg/dL, an abnormal bone marrow biopsy and anemia (see The Patients Lab Findings).

Three characteristics identify WM: an immunoglobulin M concentration greater than 3,000mg/dL, malignant lymphoplasmacytic cells in the bone marrow and anemia.
To monitor for risk of developing hyperviscosity, IgM levels in the blood are measured, as well as the patients average serum viscosity level. The average serum viscosity level for symptomatic patients is 4 centipoises (normal: 1.0cP to 1.8cP). Most people become symptomatic when the viscosity is between 5cP and 8cP, and all patients are symptomatic when serum viscosity reaches a level greater than 8cP.1,8,10
Our patients highest level of serum viscosity was 3.1cP, which correlates with his being largely asymptomatic. Other findings that are non-diagnostic include an elevated erythrocyte sedimentation rate (ESR) and thrombocytopenia, neither of which had any clinical impact.6
Patients with WM are often asymptomatic, but when symptoms arise, they usually include weakness, fatigue, epitaxis, visual symptoms, hepatomegaly, splenomegaly and lympadenopathy.1-3 Anemia is the symptom most commonly present at the diagnosis, and it is characterized by a decrease in the quantity of red blood cells, a decrease in the amount of hemoglobin or both.10 As mentioned before, our patient was anemic; both his red blood cell count and his hemoglobin were lower than normal.
Some 30% of patients with WM may experience a number of different ocular findings.8 The most common is mid-peripheral retinal hemorrhaging secondary to the hyperviscosity syndrome.3,8 As the blood becomes more viscous, the larger vessels may also become affected and can lead to a retinal vein occlusion.8 As a result, the retinal vessels may become tortuous and dilated.7,16 There are also documented cases of corneal crystals (IgM deposits), diplopia (due to mass effect of plasmacytomas), ciliary body cysts, papilledema (elevated cranial pressure due to plasma cell tumors) and serous macular detachments, all of which are very rare.7,8,10
Because WM is a rare condition, there really is no standard treatment. In the early stages of WM, the patients are observed carefully, and treatment, such as plasmapheresis, is aimed at treating the symptoms only.11-17 Plasmapheresis provides rapid relief by removing intravascular IgM and quickly decreasing any hyperviscosity.7 For advanced cases, chemotherapy has been the first line of treatment, and patients usually need to undergo at least six months of therapy because of the indolent nature of WM.3,7 In situations where chemotherapy has failed, a few patients have undergone a splenectomy. The presence of mild anemia and high IgM levels (greater than 30g/L) may predict the need to start treatment within one year from diagnosis.11-13
In a case where vessel tortuosity and retinal hemorrhaging is seen, the common differential diagnoses, such as diabetes, hypertension or retinal vein occlusions, need to be ruled outalong with some of the less common causes such as anemia, leukemia, polycyethemia or multiple myeloma.
Retinopathy secondary to anemia commonly includes intraretinal hemorrhaging, cotton-wool spots and venous tortuosity, and this can be ruled out via a routine blood test. Many WM patients are also anemic, however, so the retinopathy may result from a combination of the two conditions.
As previously mentioned, WM is only one cause of hyperviscosity syndrome; therefore, other differentials would need to be ruled out, such as chronic leukemia, polycyethemia vera or multiple myeloma with the help of blood tests.
The mortality rate due to Waldenstrms macroglobulinemia is very low, and many patients survival rates are surpassing textbook predictions of five to seven years.2 There have been reports of survivors with WM who live for 25 years after diagnosis; many are told that they are more likely to die with WM than from it.2
Ten years after diagnosis of WM, our patient is doing remarkably well. With the exception of mild hyperviscosity retinopathy, he is basically asymptomatic. Considering the history of prior treatment, initiated soon after diagnosis in the absence of any symptoms, no treatment other than observation was warranted.
1. Gertz M, Fonseca R, Rajkumar S. Waldenstrms Macroglobulinemia. The Oncologist 2000 Feb;5(1):63-7. Available at: http://theoncologist.alphamedpress.org/cgi/content/full/5/1/63 (Accessed January 7, 2007).
2. International Waldenstrms Macroglobulinemia Foundation. What is Waldenstrms Macroglobulinemia? Available at: www.iwmf.com/whatiswm.htm (Accessed March 21, 2007).
3. Menke MN, Feke GT, McMeel JW, et al. Hyperviscosity-related retinopathy in Waldenstrms Macroglobulinemia. Arch Ophthalmol 2006 Nov;124(11):1601-6.
4. Waldenstrm J. Incipient myelomatosis or essential hyperglobulinemia with fibrinogenopenia: a new syndrome? Acta Med Scand 1944;117:216-22.
5. Cutler C. Waldenstrms Macroglobulinemia. Medical Encyclopedia. 2006 Sep 20. Available at: www.nlm.nih.gov/medlinepluc/ency/article/000588.htm (Accessed January 7, 2007).
6. Virtual Blood Centre. Waldenstrms Macroglobulinemia. 2006 July 14. Available at: www.virtualbloodcentre.com/diseases.asp?did=469 (Accessed January 7, 2007).
7. Albert J, Murtha T. Principles and Practice of Ophthalmology. 2nd ed.
8. Pilon AF, Rhee PS, Messner LV. Bilateral, persistent serous macular detachments with Waldenstrms macroglobulinemia. Optom Vis Sci 2005 Jul;82(7):573-8.
9. National Cancer Institute. Waldenstrms Macroglobulinemia: Questions and Answers. July 13, 2005. Available at: www.cancer.gov/cancertopics/factsheet/sites-types/wm (Accessed January 7, 2007).
10. Pinna A, Dore S, Dore F, et al. Bilateral optic disc swelling as the presenting sign of Waldenstrms macroglobulinemia. Acta Ophthalmol Scand 2003 Aug:81(4):413-5.
11. Dimopoulos MA, Merlini G, Leblond V, et al. How we treat Waldenstrms macroglobulinemia. Haematologica 2005 Jan;90(1):117-25.
12. Alexanian R, Weber D, Delasalle K, et al. Asymptomatic Waldenstrms macroglobulinemia. Semin Oncol 2003 Apr;30(2):206-10.
13. Cesana C, Miqueleiz S, Bernuzzi P, et al. Smouldering Waldenstrms macroglobulinemia: factors predicting evolution to symptomatic disease. Semin Oncol 2003 Apr;30(2):231-5.
14. Avashia JH, Fath DF. Bilateral central vein occlusion in Waldenstrms macroglobulinemia. J Am Optom Assoc 1989 Sep;60(9):657-8.
15. International Waldenstrms Macroglobulinemia Foundation. Questions and Answers. 2003. Available at: www.iwmf.com/whatiswm.htm (Accessed January 8, 2007).
16. Albert J, Loewenstein J. Principles and Practice of Ophthalmology. 2nd ed.
17. Chen CI. Treatment for Waldenstrms macroglobulinemia. Ann Oncol 2004 Apr:15(4):550-8.
