 History
History
A 14-year-old black female presented complaining of blurred vision at distance. Her ocular history was noncontributory.
Her systemic history is remarkable for right-sided hearing loss. She said her mother and sister have the same dysfunction. She reported taking no medications and denied having allergies of any kind.
Diagnostic Data
Her best-corrected visual acuity through habitual myopic correction was 20/25 O.U. at distance and 20/20 O.U. at near. Extraocular muscle movements were normal. There was no evidence of an afferent pupillary defect. Confrontation fields were full to finger counting O.U. Refraction uncovered negligible changes, revealing -0.25D O.D. and -0.50D O.S.
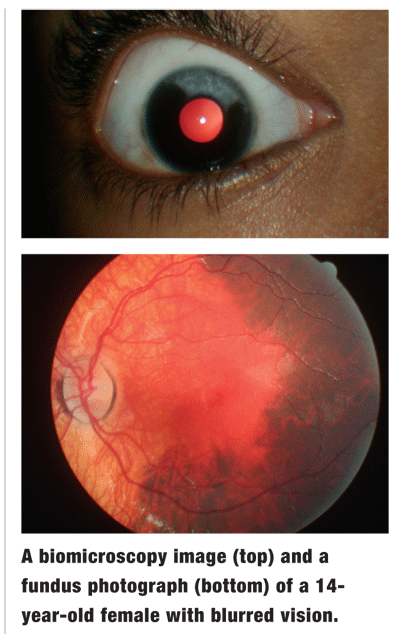
The pertinent biomicroscopic findings are illustrated in the top photograph. IOP measured 14mm Hg O.U. Pertinent findings from the dilated fundus exam are demonstrated in the bottom photograph. Additional testing included threshold automated visual fields and photodocumentation.
 Your Diagnosis
Your Diagnosis
How would you approach this case? Does this patient require any additional tests? What is your diagnosis? How would you manage this patient? What is the likely prognosis in this case?
Thanks to Christopher M. Brennan, O.D., of
This patient exhibits the classic ocular pigmentary changes associated with Waardenburg syndrome (WS).
Dutch ophthalmologist Petrus Johannes Waardenburg first described the syndrome in 1951.1-5 WS is a genetic condition inherited through autosomal dominant transmission.1-5 It demonstrates variable penetrance with no predilection for race or sex.1-5 It is caused by point mutations of single-base-pairs in the PAX3 and MITF genes.4 A first-degree relative of an affected individual often will have WS as well.
Since Dr. Waardenburg first described the condition, four distinct types of WS have been discovered.4,5 Types I and II occur most often.4 All four types share common major phenotypic findings, such as heterochromia iridis, bright blue eyes, depigmented head and/or facial hair, and congenital sensorineural hearing loss.1-5 Affected individuals also share some minor characteristics, including premature graying of hair, synophrys (eyebrow extending across the bridge of the nose) and hypoplastic nasal alae.5
WS types I and III are commonly grouped together, as are types II and IV. Types I and III also demonstrate dystopia canthorum, a lateral displacement of the inner canthi; this does not affect the interpupillary or inter-outercanthal distances.4,5 This is an important finding, as dystopia canthorum can promote pseudostrabismus.5 Another distinction between types I and III: Type III presents with hypoplastic upper limbs.5
Types II and IV differ in that patients with type IV also have Hirschsprung disease; a congenital disease of the colon that causes chronic constipation.5
In one case series, 59% of WS patients showed iris pigmentary disturbances.2 Of these, 10% also had fundus pigmentary changes.2 The iris changes can be unilateral or bilateral, producing sectoral pigmentary alterations.2 The fundus pigmentary changes usually occur in the same eye in which the iris changes occur.1
The ocular pigmentary changes, though dramatic, are not detrimental to the vision.5, Some patients may experience photophobia. However, visual function remains intact, with good visual acuity and normal responses on electroretinogram (ERG) and visual evoked potential (VEP).2 In this case, the blurry vision was not actually partial to the more global discovery.
Associated hearing loss is the most significant and debilitating characteristic of WS. One hypothesis is that melanocytes are responsible for both the development of the cochlea and Cortis organ during fetal development. WS does not allow for these melanocytes to mature, which results in hearing loss.5 Although often described as congenital deafness, the resulting hearing loss can occur at any age.5
Patients who have WS can benefit from a referral to an audiologist. Additionally, any individual suffering from WS should be made aware that up to 50% of his or her future offspring will likely have similar auditory problems. Finally, genetic counseling is appropriate for this patient.
1. Goldberg MF. Waardenburgs syndrome with fundus and other anomalies. Arch Ophthalmol 1966 Dec;76(6):797-810.
2. Delleman JW, Hageman MJ. Ophthalmological findings in 34 patients with Waardenburg syndrome. J Pediatr Ophthalmol Strabismus 1978 Nov-Dec;15(6):341-5.
3. Nork TM, Shihab ZM, Young RSL, et al. Pigment distribution in Waardenburgs syndrome: a new hypothesis. Graefes Arch Clin Exp Ophthamol 1986; 224(6):487-92.
4. Pardono E, van Bever Y, van den Ende J, et al. Waardenburg syndrome: clinical differentiation between types I and II. Am J Med Genet A 2003 Mar 15;117(3):223-35.
5. Kahn A. Waardenburg Syndrome.
