Review Education Group | Review's Continuing Education
Dozens of opportunities to earn CE credit are available through our publications and live events. Print CE courses are displayed below. Information on live events can be found here.
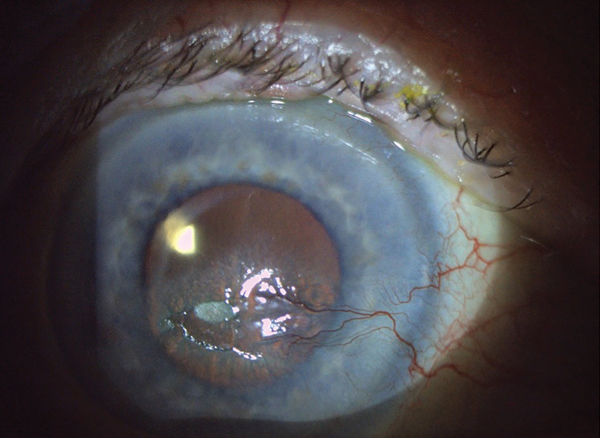
Corneal Pain Presentations: Causes and Interventions
Get up to speed on the basis and manifestations of neuropathic as well as neurotrophic changes.
By Daniel Grangaard, OD, William Hileman, OD, and Steve Njeru, OD
Review of Optometry CE, April 2024
Optometric Study Center: Courses published monthly in Review of Optometry and Review of Cornea & Contact Lenses, offering either 1 or 2 hours of CE credit. Processing fees apply. Exam valid for credit up to three years from release date.
Supported CE: Courses offered without exam fee, supported by an independent educational grant from a commercial interest. Typically valid for credit for one year from release date; longer active terms may exist for some exams.
Archived CE: Courses have expired but are displayed for reader interest. No credits available.



