A 65-year-old white male inmate presented at a state correctional facility’s eye clinic with a complaint of “bugs” in the vision of his left eye. This had been happening for about three weeks; he denied photopsia. The patient’s systemic history was significant for hypertension, heart problems (having had two valve replacements) and type 2 diabetes that was controlled with oral medications.
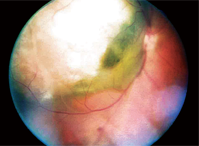
1. A fundus photograph of our patient’s right eye.
His ocular history was significant for “long-standing” blindness of his right eye.
Upon further questioning about this long-standing blindness, we learned that, 15 years earlier, the patient had seen a retinal specialist, who told him he had a “freckle” in his eye. He reported that he had lost his vision in the right eye about 10 years ago, but hadn’t seen a specialist since that visit 15 years ago.
Diagnostic Data
Upon examination, the patient’s visual acuity was light perception only O.D., and 20/25 O.S. Pupils measured 4mm O.D. and 3mm O.S. and were both round and reactive, with a 3+ afferent defect O.D. Anterior segment evaluation was remarkable for nuclear sclerotic cataracts. We performed a dilated fundus examination, and found that the patient’s chief complaint was due to a posterior vitreous detachment O.S., which caused dense, central vitreous syneresis. We found no retinal breaks or anything else of clinical significance in that eye.
Dilated fundus examination of his right eye revealed a large elevated lesion, approximately 12 disc diameters in size, extending from—and including—the temporal optic disc, past the temporal macula and beyond the superior arcade. It was gray-white in color with what appeared to be some fluid content in the inferior-nasal aspect (figure 1).
Diagnosis
We made a provisional diagnosis of choroidal melanoma with serous retinal detachment, and documented it with fundus photos.
Treatment and Follow-Up
We referred the patient to the facility’s staff ophthalmologist for further evaluation and treatment. About two weeks later, the ophthalmologist saw him and took a fluorescein angiogram as well as an A- and B-scan ultrasound. The interpretation reports for these procedures were fairly rudimentary; they stated that the A- and B-scans revealed a “solid mass,” and the fluorescein showed that the mass was filled with dye early and then slowly disappeared. After these studies, the ophthalmologist posed differentials of malignant melanoma or disciform macular degeneration. He referred the patient to a retinal specialist, who saw him just days after his ophthalmology consultation.
The retinal specialist immediately diagnosed a choroidal melanoma and sent the patient to an oculoplastics specialist that day to discuss treatment. Diagnostic testing results from the retinal specialist and ocuplastics specialist were not available to us in the patient’s record. An abdominal CT scan with contrast and a chest X-ray were also ordered.
The patient and oculoplastics specialist decided on treatment with enucleation, and the patient was scheduled for surgery the following week. The enucleation was successful; however, the patient unfortunately experienced difficulty with the anesthesia and died two days after the enucleation. The abdominal CT and chest X-rays had not yet been completed.
Discussion
Choroidal melanomas are relatively rare, with an incidence of approximately five to six cases per one million people, which equates to about 1,400 cases in the United States each year.1,2 They are found mostly in adults (with the peak around age 55), generally are not familial, and show a slight male predilection for most age groups.1,3,4 They occur mostly in light-skinned persons with blue or green irides, and are rarely found in blacks or Asians.1,4
Patients with choroidal melanomas are often asymptomatic, but may present with decreased vision, visual field defects, floaters, photopsias or, in rare instances, pain.4,5 If pain does occur, it is usually a result of secondary glaucoma or tumor necrosis; choroidal melanomas also can cause pain by impinging on underlying posterior ciliary nerves, but this seldom occurs.3,5
These lesions usually are elevated and may appear mottled, dark brown, dull-gray, gray-green or yellow (amelanotic).4-6 They may assume a mushroom or dome shape with congested blood vessels within the tumor—this configuration represents the 20% of choroidal melanomas that erupt through Bruch’s membrane and the retinal pigment epithelium (RPE).1,4,5Choroidal melanomas often display an abrupt elevation from the choroid, subretinal fluid, orange pigmentation over the lesion’s surface and growth over time.4 Subretinal fluid with resultant underlying serous retinal detachment results from RPE breakdown. These serous detachments often shift, and may appear to contain blood if the tumor has traversed Bruch’s membrane.5
Overlying orange pigmentation is lipofuscin; this pigment is composed of proteins, lipids and small chromophores, and it accumulates in the RPE as a result of cell degeneration and incomplete digestion of the photoreceptor outer segments.6,7 Lipofuscin is not specific to melanomas; it may also be associated with choroidal nevi or other benign choroidal tumors. However, lipofuscin is much more commonly seen with melanomas than with benign counterparts.5
Other possible signs associated with choroidal melanomas include vitreous hemorrhages or pigmented vitreous cells, drusen on the surface of the tumor, choroidal neovascular membranes, or even proptosis if the tumor invades the orbit.4
Differential Diagnoses
There are a plethora of differential diagnoses for melanotic and amelanotic choroidal melanomas, which vary on the prognostic continuum of severity.
• Choroidal nevi are a major differential. They are common, benign melanocytic tumors and are found in approximately 2% to 6.5% of the white population.8-10 Nevi usually are slate-gray and relatively flat (less than 2mm thickness), although there is significant size overlap between small melanomas and larger nevi.8,9 Like choroidal melanomas, they also may show overlying drusen or lipofuscin (figure 2). Statistically, of every 500 choroidal nevi, one will undergo malignant transformation if followed for 10 years; the estimated annual rate of malignant transformation is one in 8,845.6,9
There are multiple known risk factors for such transformation (see “Malignant Transformation.”).4,8 The most important appears to be initial nevus thickness of greater than 2mm, but a large base diameter (greater than 7mm) also suggests premalignancy of the nevus.8,9 The absence of drusen is a good prognostic indicator.7
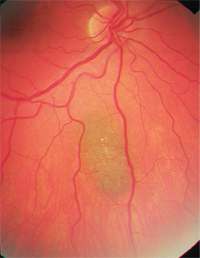
2. Choroidal nevus with overlying drusen.
Whereas choroidal melanomas tend to grow relatively rapidly, choroidal nevi may enlarge slowly over a period of several years, not necessarily indicating malignant transformation. Such non-malignant growth is more common in younger patients and tends to stabilize with age.11 Thus, slow growth of choroidal nevi is not invariably a sign of malignancy, especially in younger patients without other risk factors.11
Interestingly, pigmented choroidal lesions with none of the above risk factors have a 3% chance of growth in five years; such lesions are usually choroidal nevi.9 Presence of one of the above factors carries a 38% chance of growth, and more than a 50% chance of growth exists if two or more risk factors are present.9 The relative risk of growth climbs from 1.9 times to 27.1 times for the presence of one vs. all five risk factors.9
• Choroidal metastasis refers to a tumor that has spread to the choroid via hematogenous routes from a primary malignancy elsewhere in the body. Thus, they are not primary tumors like choroidal melanoma—most often, they are metastases from breast or lung cancer. These lesions usually appear dome-shaped and creamy-yellow in color, and often induce retinal detachments. Choroidal metastases are often bilateral or multifocal and do not appear mushroom-shaped, unlike amelonatic melanomas.1
• Congenital hypertrophy of the RPE presents as single or multifocal, darkly-pigmented, flat lesions, often with hypopigmented lacunae. They are benign, usually do not change with time, and require no treatment.1
The resultant serous retinal detachment and retinal elevation secondary to exudative age-related macular degeneration (AMD) poses another differential for choroidal melanoma. AMD may show subretinal hemorrhaging, lipid or turbid exudation, dirty-gray or yellow macular edema, choroidal folds, pigment epithelial detachments or disciform scarring.1 Fluorescein angiography aids in differentiating between these conditions.
• Melanocytomas are darkly pigmented and found on or around the optic disc (figure 3). Unlike melanomas, they are congenital and often occur in individuals with dark pigments. They usually are inactive, but may grow and rarely develop into melanomas.1
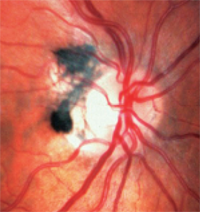
3. Melanocytoma are darkly pigmented and found on or around the optic disc.
• Choroidal hemangiomas are benign dilations of choroidal blood vessels and are often associated with Sturge-Weber syndrome. They appear elevated and are red-orange in color. Like choroidal melanomas, they may induce serous retinal detachments.1
• Choroidal osteomas are yellow-orange placoid masses. Interestingly, they are composed of mature bone tissue. They may allow choroidal neovascularization and subretinal bleeding to develop. Very characteristic features make them easy to differentiate from choroidal melanomas using ultrasonography or CT scan.1
• Additional differentials for amelonotic or melanotic choroidal melanomas include choroidal detachment, lymphoma, metastatic carcinoma, subretinal or sub-RPE hematoma, localized suprachoroidal hematoma, nodular posterior scleritis, reactive hyperplasia of RPE or massive gliosis of the retina.4,5
Additional Testing
Various instruments may assist in the diagnosis of choroidal melanomas.
A- and B-scan ultrasonography not only aids in diagnosis, but also may provide more accurate measurements of the tumor. A-scan usually reveals low internal reflectivity within the tumor; oscillation in height of the echoes within the lesion may correspond with the patient’s pulse, which indicates the presence of intralesional vascularity.1,5
B-scan shows a solid mass with an acoustically bright anterior aspect with internal and basal darkness; the cross-sectional shape typically is bi-convex, but may appear mushroom-like. Choroidal excavation and orbital shadowing may also be seen.1,5
Fluorescein angiography typically shows hyperfluorescence of the tumor’s vessels and diffuse late staining.1 However, the fluorescein pattern depends on the tumor’s size, shape, pigmentation, integrity of the RPE and whether there is a corresponding serous retinal detachment, among other variables.5 Fluorescein angiography does not yield pathognomonic signs of choroidal melanoma.3
In this particular patient, choroidal melanoma was diagnosed based on fundoscopic examination, A- and B-scan ultrasounds, and fluorescein angiography (without biopsy).
This patient’s clinical presentation alone was highly suggestive––basically unequivocally––of choroidal melanoma. He possessed four of the five aforementioned risk factors for malignancy: thickness > 2mm, subretinal fluid, symptoms/visual loss, and a location < 3mm from the optic disc. Though the dimensions of the melanotic lesion based on ultrasonography are not available, it was clearly thickened more than 2mm and had a basal diameter larger than 7mm (the widely accepted upper limits of benign nevi).8,12
Additionally, the mass exhibited blatantly invasive features, such as encroachment onto the optic disc.8,12
Management and Prognosis
When a suspicious ocular mass is found, it is important to ask the patient whether they have had any ocular surgery or trauma; a history of cancer; or any systemic symptoms of cancer, such as anorexia, weight loss, general fatigue, malaise or illness. While 98% of patients with choroidal melanomas do not have metastatic disease detectable at the time of diagnosis, metastasis must be ruled out.5 This would most appropriately be handled by an ocular oncologist, so such a referral should be made.
Tests include a complete blood count, liver enzymes, abdominal CT, MRI or ultrasound, and a chest X-ray.4,5 Several treatment options are available for choroidal melanomas, but many have high risks involved; therefore, the treating practitioner must carefully weigh many variables when selecting the appropriate treatment modality for each particular patient. A few factors to consider are tumor size and location, metastasis status, the visual status of both the affected and unaffected eyes, and the patient’s age and overall health.5 Depending on these factors, observation may be a viable management plan if the patient has serious concurrent medical issues, but generally is not advised.
Malignant Transformation
Risk factors for malignant transformation of choroidal nevi include:4
• Thickness > 2mm.
• Subretinal fluid.
• Presence of symptoms.
• Prominent orange pigment overlying the lesion.
• Location < 3mm from the optic disc.
*If two or more factors are present, the lesion is likely a choroidal melanoma.
One very aggressive treatment is enucleation, but it comes with significant risks. Half of the patients who are treated with enucleation eventually die of metastatic melanoma. This invasive treatment option is more often discussed if the affected eye is blind, painful, shows optic disc involvement, or if the tumor is very large.5
Most small choroidal melanomas are treated with locally destructive therapies, such as thermotherapy, radiotherapy or irradiation.12 Various types of radiation may be used as treatment.4,5 The most common is plaque brachytherapy, which utilizes a radioactive plaque that is sutured on the surface of the globe exterior to the tumor.
This is more commonly attempted with smaller tumors that are 3 disc diameters or more away from the disc and fovea. Approximately 10% to 15% of patients treated in this way experience local tumor relapse after treatment. Post-treatment, the patient’s vision usually remains the same as it was before treatment, but there is a chance that it may improve. However, vision may be subsequently reduced due to secondary effects, such as radiation retinopathy, optic papillopathy, cataracts or neovascular glaucoma.
Photocoagulation may be attempted for small tumors (< 3mm thickness, < 7mm basal diameter).4,5 Like photocoagulation for any other reason, a permanent scotoma will result in the photocoagulated areas. Other laser treatments can also be used, including transpupillary thermotherapy, which utilizes a low-power, long-duration infrared laser.5,12 This technique may be used in conjunction with plaque radiotherapy, but has not shown a significant improvement in local tumor control.13
Other, less common treatments include local resection, photodynamic therapy or cryotherapy.4,5 Often, multiple treatments are used as part of a combination approach.
Sadly, the prognosis for patients with choroidal melanoma often is poor. Despite treatment, 30% to 50% of patients eventually develop metastatic disease; this occurs preferentially to the liver, but also to lung, bone, skin, lymph nodes or central nervous system.3,11,14 The same proportion of patients will die within 10 years from diagnosis, usually due to metastatic spread.3,11
Once metastasized, fatality is almost certain.11 The highest incidence of metastatic detection is within a year from choroidal melanoma diagnosis, though it may not occur until years later. Several factors correlate with increased mortality rate, including larger melanoma size, anterior location, extrascleral extension, growth through Bruch’s membrane, optic nerve extension, lack of pigmentation, and aggressive cell type and/or mitotic activity.3
It is apparent that this patient did not receive adequate care at his first appointment with the optometry clinic; however, despite appropriate referrals following his second appointment, the chances of a successful outcome were reduced markedly.
Though a delay in referral of a few months may not have significantly altered outcomes in this case, this situation emphasizes the importance of thorough case history and effective doctor-patient communication. It also highlights the importance of appropriate referrals and additional work-ups, regardless of the “long-standing” nature of a condition.
While the prognosis for patients with choroidal melanoma may seem bleak, eye care professionals must institute appropriate treatment as soon as they discover such a lesion to improve the patient’s chances of having positive secondary outcomes, including preservation of vision.
Dr. Weidmayer practices with a group of optometrists at Eye Center of Lenawee, P.C., in Adrian and Brooklyn, Mich.
1. Spaide RF. Diseases of the Retina and Vitreous. 1st ed. Philadelphia: W.B. Saunders; 1999:262-65.
2. Margo CE. The collaborative ocular melanoma study: an overview. Cancer Control. 2004 Sep-Oct;11(5):304-9.
3. Garcia-Valenzuela E, Pons ME, Puklin JE, Davidson CA. Choroidal Melanoma EMedicine Ophthalmology. Medscape Reference. June 24, 2009.
http://emedicine.medscape.com/article/1190564-overview. Accessed August 17, 2010.
4. Choroidal nevus and malignant melanoma of the choroid. In: Ehlers JP, Shah CP (eds). The Wills Eye Manual: Office and Emergency Room Diagnosis and Treatment of Eye Disease. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2008:330-3.
5. Augsburger JJ, Damato BE, Bornfeld N. Uveal Melanoma. In: Yanoff M, Duker JS, eds. Ophthalmology. 1st ed. London: Mosby;1999:1052-63.
6. Jones WL. Ophthalmoscopy. In: Terry JE (ed). Ocular Disease: Detection, Diagnosis, and Treatment. 1st ed. Boston: Butterworth Publishers; 1984:155-57.
7. Materin MA, Raducu R, Bianciotto C, Shields CL. Fundus autofluorescence and optical coherence tomography findings in choroidal melanocytic lesions. Middle East Afr J Ophthalmol. 2010 Jul;17(3): 201-6.
8. Augsburger JJ, Correa ZM, Trichopoulos N, Shaikh A. Size overlap between benign melanocytic choroidal nevi and choroidal malignant melanomas. Invest Ophthalmol Vis Sci. 2008 Jul;49(7):2823-8.
9. Kaiserman I, Kaiserman N, Pe’er J. Long term ultrasonic follow up of choroidal naevi and their transformation to melanomas. Br J Ophthalmol. 2006 Aug;90(8):994-8.
10. Mashayekhi A, Siu S, Shields CL, Shields JA. Slow enlargement of choroidal nevi: a long-term follow-up study. Ophthalmology. 2011 Feb;118(2):382-8.
11. Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010 July;12(4):461-8.
12. Augsburger JJ, Correa ZM, Schneider S, et al. Diagnostic transvitreal fine-needle aspiration biopsy of small melanocytic choroidal tumors in nevus versus melanoma category. Trans Am Ophthalmol Soc. (2002);100:225-34.
13. Sagoo MS, Shields CL, Mashayekhi A, et al. Plaque radiotherapy for juxtapapillary choroidal melanoma: tumor control in 650 consecutive cases. Ophthalmology 2011 Feb;118(2):402-7.
14. Finger PT, Kurli M, Reddy S, et al. Whole body PET/CT for initial staging of choroidal melanoma. Br J Ophthalmol. 2005 Oct;89(10):1270-74.
