 A 25-year-old white female presented for an eye examination after she failed the required vision test for obtaining her drivers license. She reported that she had vision difficulties for as long as she could remember. She said that she wore glasses in the past, but they were of little help. Her medical, family and ocular histories were unremarkable.
A 25-year-old white female presented for an eye examination after she failed the required vision test for obtaining her drivers license. She reported that she had vision difficulties for as long as she could remember. She said that she wore glasses in the past, but they were of little help. Her medical, family and ocular histories were unremarkable.
On examination, her best-corrected visual acuity measured 20/200 O.U. at distance and near. Confrontation visual fields were full to finger counting O.U. Her pupils were equally round and reactive to light, with no afferent pupillary defect. Extraocular motility testing was normal. Her anterior segment examination was unremarkable.
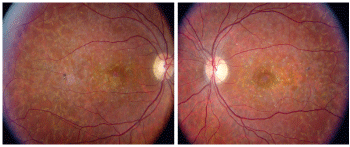
1,2. Posterior pole fundus images of our patient with 20/200 vision O.U. (O.D. left, O.S. right).
The dilated fundus exam showed moderate-sized cups, with good rim coloration and perfusion O.U. The patient had mottling of the retinal pigment epithelium (RPE) O.U. (figures 1 and 2). She also demonstrated some peculiar changes in both maculae, as well as some white spots throughout her posterior poles. The vessels and peripheral retinas were normal. A fluorescein angiogram (FA) was performed (figure 3).
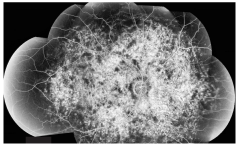
3. Widefield montage fluorescein angiogram O.D.
Take the Retina Quiz
1. How would you describe the significant findings seen on FA?
a. Marked staining and blockage of the choroidal fluorescence.
b. Blockage of the background choroidal fluorescence.
c. Hyperfluorescence of the macula in a bulls-eye pattern.
d. Milky way in the night sky staining pattern.
2. What is the correct diagnosis for this patient?
a. An occult retinal dystrophy.
b. Familial drusen.
c. Stargardts disease (SD).
d. Cone dystrophy.
3. Whats the conditions etiology?
a. Nonspecific degenerative process of the retina.
b. Lipofuscin storage disease.
c. Congenital loss of cone receptors.
d. Abnormal complement factor H (CFH) gene.
4. Which gene is linked to this patients condition?
a. CFH.
b. Rhodopsin (
c. XLRS1.
d. ABCA4.
For answers, see below.
Discussion
Our patient has a fairly advanced case of Stargardts disease. On clinical examination, extensive atrophy and mottling of the RPE is seen throughout both retinas. Extensive atrophic changes are evident in both maculae, and on careful inspection, the classic beaten-bronze appearance is still somewhat preserved.
SD is an autosomal recessive disorder that is present in about one in 10,000 people.1 It is the most common hereditary macular dystrophy.1,2 The typical age of initial presentation ranges from the teens to early 20s; however, cases with a later onset have been reported.
The clinical findings in SD vary among patients. The macula may appear normal or show atrophic RPE changes that resemble the classic beaten-bronze appearance, as seen in our patient. Patients may have macular atrophy with or without scattered yellow-white flecks, or they may present with flecks in the absence of macular atrophy.
These yellow-white flecks may appear throughout the fundus, and can vary in size and shape. Some exhibit a pisciform (fish-like) or triradiate pattern. Over time, the flecks can disappear, which results in RPE/choriocapillaris atrophy; new flecks may appear at any time. The flecks increase in number as the disease progresses.
Had our patient been in her 60s or 70s, we could easily have mistaken the clinical picture for age-related macular degeneration, as the white spots in the posterior pole look like drusen. However, these spots are areas of localized accumulation of lipofuscin.
In cases of SD, the fundus appears vermillion, or deep red, in color. This is because SD is a lipofuscin storage disease, which causes a loss of choroidal detail.
Lipofuscin is a ubiquitous, insoluble material that is present in granules in the RPE cell, with a characteristic UV-excitable fluorescence. It is the product of hydrolysis of vitamin A aldehyde and phosphatidylethanolamine.
In SD, lipofuscin accumulates at abnormally high concentrations within the RPE and does not permit observation of the choroid below the RPE.3
Also, because of increased levels of lipofuscin, the fluorescein staining pattern will show blockage of the background choroidal fluorescence, which results in a quiet choroid and is highly diagnostic.
Our patient has a quiet choroid, but it is difficult to recognize due to the widespread atrophy of the RPE throughout the posterior pole. Once outside the areas of atrophy, however, the choroid can be seen in the fluorescein montage.
In recent years, researchers have discovered that the ABCA4 gene is responsible for SD. The ABCA4 gene codes an energy-dependent transmembrane protein that is localized in the discs of the rod and cone outer segments. The protein is a member of the ATP-binding cassette (ABC) transporter superfamily and functions in the transport of vitamin A in the visual cycle.2
Unfortunately, there is no treatment for SD. In many academic settings, patients with confirmed SD are entered into a Stargardts disease registry, and their blood samples are sent to one of several sites in the
We explained the condition to our patient and informed her that she did not meet the vision requirements necessary to obtain a drivers license. We then recommended a low vision examination.
Retina Quiz Answers: 1) a, 2) c, 3) b, 4) d.
1. Blacharski PA. Fundus Flavimaculatus. Retinal Dystrophies and Degenerations.
2. Deutman, AF, Hoyng CB, van Lith-Verhoeven JJC. Macular dystrophies. In: Ryan SJ (ed). Retina, Vol. II: Medical Retina, 4th edition.
3. Marmorstein AD, Marmorstein LY, Sakaguchi H, Hollyfield JG. Spectral profiling of autofluorescence associated with lipofuscin, Bruchs membrane, and sub-RPE deposits in normal and AMD eyes. Invest Ophthalmol Vis Sci 2002 Jul;43 (7):2435-41.
