Several months ago, a 63-year-old patient with glaucoma presented for a complete eye examination as a new patient. He reported being followed for glaucoma for the past 10 years and being diagnosed with mild hypertension three years prior.
He said his hypertension was being successfully controlled with medication. As for his glaucoma treatment, he reported having tried a number of different medications over the past years and was currently taking Travatan (travoprost ophthalmic solution, Alcon) q.h.s. in both eyes. He denied any side effects from the glaucoma medication and described good compliance with his dosing schedule.
His best-corrected visual acuity was 20/40 O.D. and 20/50 O.S. with no improvement on pinhole, and his intraocular pressures measured 13mm Hg O.D. and 14 mm Hg O.S. with Goldmann tonometry.
Dilated fundus examination revealed pale optic nerve heads with minimal cupping in both eyes. Humphrey visual fields (24-2) revealed dense superior defects in both eyes.
We were able to obtain this patients complete records from his previous optometrist. These records confirmed our patients history. Of particular interest: His visual fields the day he was diagnosed with glaucoma were nearly identical to what was found the day he presented as a new patient. Subsequently, this patient was correctly diagnosed with non-arteritic anterior ischemic optic neuropathy (NAAION).
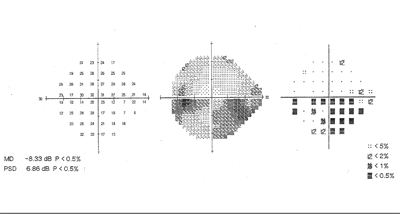 |
|
SITA Standard 24-2 Humphrey visual field in a patient with NAAION. Inferior nasal visual field scotomas are the most common defect seen in these patients. |
NAAION
NAAION is the second most common optic neuropathy. It typically occurs in patients past age 50, with men and women affected equally.1,2
NAAION is an ischemic process that affects the paraoptic branches of the short posterior ciliary arteries. Recent studies show that infarction primarily occurs deep in the retrolaminar area of the nerve head and only occasionally extends to the laminar and prelaminar areas.3 This infarction occurs in the absence of any compression, inflammation or demyelination.4
 |
|
A significant association has been found between oral erectile dysfunction drugs, such as Viagra, and NAAION in patients who have a history of myocardial infarction or hypertension. |
There is a wide variation of presenting vision in NAAION. Dyschromatopsia appears to be proportional to the amount of vision loss. Visual acuity is better than 20/64 in 50% of NAAION patients.6 Visual field loss accompanies and commonly results in nasal and altitudinal defects inferiorly more often than superiorly.7
In the acute phase of NAAION, the optic nerve may demonstrate sectoral to complete optic disc edema with splinter hemorrhages. Some researchers believe that disc edema may precede the vision loss and that swelling exacerbates the ischemic attack by further limiting blood flow.1,3 The hemorrhages tend to resolve within the first several weeks, followed by the acute edema. As the edema subsides and ganglion loss occurs, optic atrophy may appear as early as one month, leaving sectoral to diffuse nerve head pallor and/or excavation.8
Evaluation of the fellow eye typically reveals a disc at riska small, crowded disc with minimal cupping. This small disc coupled with coexisting vascular disorders, such as hypertension, diabetes, hyperlipidemia and/or arteriosclerosis, significantly increases the risk of NAAION in this eye.9 Buried optic disc drusen also increases the risk of NAAION in the fellow eye, crowding an already congested disc.10 Still, only 18% of patients have fellow eye involvement.3
NAAION is a diagnosis of exclusion.4 Therefore, it is essential to eliminate all other possible etiologies, especially AAION in patients past age 60, optic neuritis in patients younger than age 40, and the remaining optic neuropathies.
If a male patient presents with signs of NAAION and has a history of myocardial infarction or hypertension, ask him about use of oral medications for erectile dysfunction. A significant association was found between these medications and NAAION in patients who fit this profile.11 Attempt to identify these at-risk patients, and educate them on the potential ocular complications associated with the use of these drugs.11 (For more on these agents, see Erectile Dysfunction Drugs and the Eye, January 2006.)
Aside from treating any underlying systemic disease, no current definitive treatment is beneficial to patients who have NAAION. Some 43% of patients visual acuity spontaneously improved by three or more lines within the first six months, the Ischemic Optic Neuropathy Decompression Trial (IONDT) found.5 However, the IONDT found corticosteroid use to be ineffective, and the study was abandoned when optic nerve sheath fenestration failed to show long-term benefits and actually worsened many patients prognosis.12
AAION
AAION, an inflammatory disorder of the arteries that supply the optic nerve, accounts for 12% of all ischemic optic neuropathies.13 This condition occurs more often in women, and patients are generally older (with an average age of 75 at onset) than those who have NAAION.13,14 Vision loss is typically severe, sudden or rapidly progressive, with most patients having acuity of 20/200 to no light perception (NLP).14
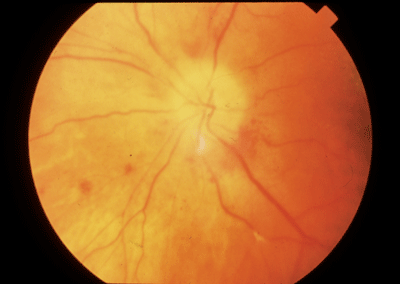 |
|
AAION is an inflammatory disorder of the arteries that supply the optic nerve. |
AAION presents as a relative afferent pupillary defect along with a pale or chalky-white, edematous disc with splinter hemorrhages. With time, the nerve becomes atrophic as the edema and hemorrhages resolve, and the visual prognosis remains very poor. In measurable eyes, altitudinal or central visual field defects are common. A palpable and tender temporal artery may also be visible.13-14 What constitutes a measureable eye depends on the type of visual field used (Goldmann vs. automated) and is very patient dependent (final acuity, scotomas, patient reliability, etc.).
|
FAST FACTS |
Ocular complications of GCA include:15
Transient monocular blindness (TMB) episodes, which occur in 2% to 19% of cases.
Ocular motility disturbances, 2% to 43% of cases. These cases typically involve cranial nerve III without pupillary involvement.
Retinal artery occlusion, 5% to 10% of cases. This most frequently involves the central retinal artery.
AAION, which is the most common cause of severe vision loss in patients who have GCA.
GCA mortality rates can be as high as 20% in untreated patients.1,3 So, you should assume that any elderly patient you suspect has AAION, has it secondary to GCA, and refer these patients for an emergency neuro-ophthalmology consultation to initiate treatment and laboratory workup.
This workup should include a Westergren erythrocyte sedimentation rate (ESR) in conjunction with a C-reactive protein test.15-17 Neither ESR nor CRP is specific for GCA, but both can indicate acute inflammatory activity. ESR is typically grossly elevated in patients who have AAION (at least 50mm/hr).16,17 However, 15% to 20% of AAION patients have a normal ESR, so this test alone is not definitive.15-17 Some studies report that ESR and CRP together have a 97% sensitivity for diagnosing GCA.15 However, in any symptomatic or high-risk AAION patient, a superficial temporal artery biopsy (TAB) should be performed.17
Elderly patients who experience sudden vision loss or who exhibit symptoms of GCA should be assumed to have GCA and treated as such, until proven otherwise. Treatment consists of high-dose corticosteroid therapy, typically administered intravenously. Methylprednisolone 250mg every six hours for 12 doses is common, and oral maintenance doses remain high (80 to 120mg/ day) until symptoms disappear or the ESR is reduced.13 The treatment course is long, and tapering must be extremely slow, often over one to two years, to prevent reactiviation. Even with therapy, however, vision improvement is minimal and only occurs in 15% to 34% of patients.17
While AAION is fairly synonymous with GCA, it can be caused by any vasculitis, such as systemic lupus or polyarteritis nodosum.8
Papilledema
Papilledema is optic disc swelling and elevation in response to increased intracranial pressure.18-21 As intracranial pressure increases, axonal transport in the optic nerve is impeded in the prelaminar area, causing the optic nerve to distend. As the nerve head swells forward into the vitreous, concurrent swelling occurs laterally, causing Patons folds (retinal folds or lines) circumferentially around the optic nerve head.19
Patients have reported transient vision obscurations or episodes of blurred vision that last two to 30 seconds; these may be prodromal symptoms of papilledema.18,19,21 Other symptoms: headaches that are worse upon waking and with postural changes, transient horizontal diplopia, mild neck discomfort, tinnitus, nausea, vomiting and dizziness.18 Visual acuity is typically normal or may be slightly decreased in patients with macular involvement.19 Patients who do not have macular involvement typically have normal vision until chronic papilledema progresses to optic atrophy. Color vision and pupils generally remain intact until chronic papilledema leads to axonal death and optic atrophy.18,19
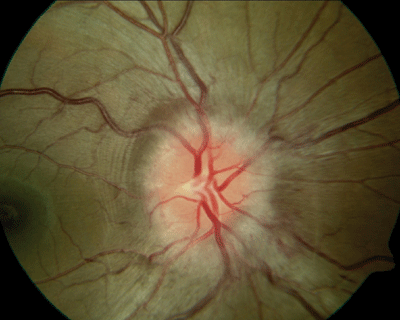 |
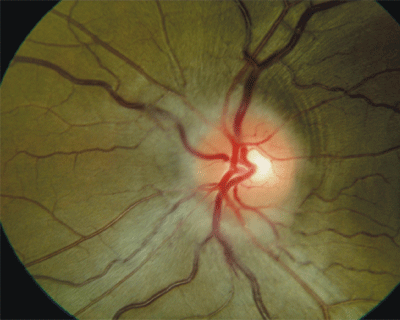 |
|
Although papilledema is a bilateral disease, it may present asymmetrically, as seen in this patient who has idiopathic intracranial hypertension. The cup has been obliterated in the right eye (above) and splinter hemorrhages are apparent. The left eye (below) is hyperemic, with some blurring of the disc edges; however, the cup is still distinguishable. Note the obvious Patons lines in both eyes. |
Patons folds lead to the classic blind spot enlargement seen on perimetric fields. Nerve bundle defects, such as arcuates, nasal steps, generalized depression, constriction and optic atrophy, are typically seen in the later stages of papilledema.19-21 Atrophic discs without viable axons do not exhibit optic nerve head edema. This gives the appearance of a unilateral condition, which may be misleading.18
Increased intracranial pressure may result from an intracranial or spinal column tumor, cerebral edema or encephalitis, epidural or subdural hematoma, subarachnoid hemorrhage, meningitis, venous sinus thrombosis (VST), drug or vitamin toxicity, or idiopathic intracranial hypertension (IIH).18-22
IIH, also known as benign intra-cranial hypertension and pseudotumor cerebri, is a diagnosis of exclusion after you eliminate all other possible etiologies. IIH is a disease of unknown etiology that primarily affects obese women of childbearing age (20 to 44 years).18 Bilateral papilledema is a hallmark sign of the disease. Some studies suggest that excess weight in the abdominal area causes a chain reaction from increased intra-abdominal pressure, eventually leading to increased intra-cranial venous pressure.23
Neuroimaging reveals unremarkable brain structure, although magnetic resonance venography (MRV) shows the narrowing of the transverse dural venous sinus, which in-creases the risk for IIH in obese female patients.24 Cerebrospinal fluid (CSF) examination is normal in patients who have IIH, although their opening pressure exceeds 250mm of water.22-26
While IIH may be self-limiting, the severe intractable headaches and progressive vision damage can be treated with diuretics, namely Diamox or Diamox Sequels (acetazolamide, Lederle.) This carbonic anhydrase inhibitor has been shown to decrease CSF production by 50%.27 To diminish symptoms (intractable headaches, dizziness, blurred vision, neck discomfort, etc.) and visual complications, start with 250mg p.o., and slowly increase the patients dose by 250mg per day until a total of 1g to 4g p.o. b.i.d. is reached or the patient becomes intolerant.
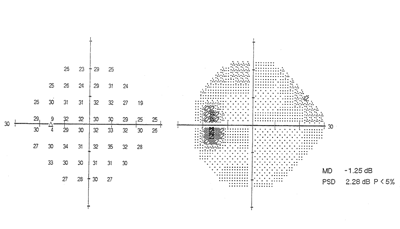 |
| Blind spot enlargement is a common visual field defect in papilledema. |
Although corticosteroids have been prescribed when papilledema is associated with inflammatory disorders, this is a controversial treatment, as well-established side effects include weight gain and water retention.27 Optic nerve sheath fenestration has proven to be a useful and effective tool for patients who have compromised vision, although it typically has no effect on the symptomatic headache of IIH.25
Lumboperitoneal and ventriculo-peritoneal shunts have been shown to be very effective in decreasing acute papilledema and in treating intractable headaches.26 More than 82% of patients reported immediate relief following the procedure.26 Still, these neurosurgeries have high failure rates, leading to increased need for revisions, meaning redoing or adjusting the procedure.26
Thus, weight loss is considered the first line of treatment for IIH.27 In one study, a 6% weight loss was associated with a significant resolution of papilledema and its accompanying symptoms.27 However, because many patients in this study lost only an average of 3.3% of their body weight, bariatric surgery may be necessary for individuals unable to achieve adequate weight loss.27
All patients who present with papilledema should undergo immediate neuroimaging to rule out a space-occupying lesion, a thorough blood workup with complete blood count to rule out infectious etiologies and blood coagulopathies, and blood pressure measurement to rule out malignant hypertension.22 Also, B-scan ultrasonography can differentiate between pseudopapilledema (in cases of optic nerve head disc drusen) and true papilledema.22
The visual prognosis for these pa-tients is typically very good if the underlying condition is addressed promptly. Regular follow-up care with dilated examination and visual field testing is important to assess and monitor patients for visual changes or deterioration. Educate patients that papilledema can lead to optic atrophy and irreversible vision damage if left untreated.18-19,27
Toxic Optic Neuropathy
Toxic, or nutritional, optic neuropathy is largely isolated to patients who have personal histories of alcohol and tobacco abuse.28 So, there is no age, sex or race predilection.28-29
Patients typically complain of a gradually increasing blurring or dimming of their central vision, and they may report dyschromatopsia.29 Visual decline is painless, progressive, bilateral and mostly symmetric; but vision typically remains measurable at 20/50 to 20/200.28,29 It rarely becomes worse than 20/400.29
Formal static or kinetic visual field testing often reveals central or cecocentral scotomas.28-30 On examination, the optic nerve may have a wide range of appearances, ranging from unremarkable to edematous with peripapillary hemorrhages in acute toxicity.29 In chronic cases, optic atrophy may be apparent with temporal pallor and nerve fiber loss greatest in the papillomacular bundle; this corresponds directly with the central or cecocentral visual field defects typically seen.28-30 Afferent pupillary defects are uncommon, as the disease is bilateral. However, both pupils are generally sluggish in their light reaction.28-30
Although several substances have a direct toxic effect on the optic nerve (see Some Common Substances Implicated in Toxic Optic Neuropathy, below), we do not know whether malnutrition and toxicity work together or independently. In most patients with toxic neuropathy and a history of chronic drug or alcohol abuse, damage stems from a nutritional deficit, specifically vitamin B12. Alcohol consumption not only decreases vitamin B12 absorption directly, but the nutritional and dietary intake tends to be poor in substance abusers, which compounds the deficiency. However, acute toxicity resulting in vision loss within hours to days has been reported with extreme alcohol ingestion, supporting a direct toxic relationship.30-32
|
Some Common Substances Implicated In Toxic Optic Neuropathy28,31,34 |
|
Amiodarone |
To determine the inciting agent, perform a thorough case history that includes questions about past and current drug or substance abuse; systemic medications; dietary intake; social background, including other signs of malnutrition; and occupational environment to rule out exposure to toxic substances. In addition, a laboratory workup with complete blood count (CBC); serum levels of vitamins B1, B12 and folate; and a heavy metal screen (e.g., lead or thallium) can pinpoint the responsible agent.35
 |
|
Toxic, or nutritional, optic neuropathy is largely isolated to patients who have personal histories of alcohol and tobacco abuse. |
Supplements of the appropriate nutrient or vitamin, or removal of the insulting toxin may reverse vision loss and field defects if identified and treated early.29-31 Follow-up should initially occur monthly and then every six to 12 months.35
Patients who have systemic conditions that require medications implicated in optic neuropathies should undergo a complete eye examination with baseline color vision and formal visual field testing and be monitored at six-month intervals. Patient education and home monitoring with an Amsler grid are also important for early intervention and management to prevent irreversible vision damage.35-36
Optic Neuritis
Optic neuritis is inflammation of the optic nerve.37 Occasionally, this inflammation occurs due to a systemic inflammatory process or infection, such as Lyme disease. More commonly, however, it occurs due to a demyelinating event and is a hallmark sign of multiple sclerosisthe most common cause of nerve fiber demyelination.37,38
Demyelinating optic neuropathy, or retrobulbar optic neuritis, presents as unilateral vision loss in younger patients, typically ages 15 to 45, who are female (77%).38 Pain that increases with eye movement, relative afferent pupillary defect and decreased color saturation in the afflicted eye are evident. Visual field defects vary greatly. Along with visual acuity changes, episodes of increased severity of vision depression and ocular pain may be associated with exercise or increased body temperature, known as Uhthoffs phenomenon.37 Although optic disc edema is seen in a few patients, often there are no ophthalmoscopic signs of inflammation.38-39
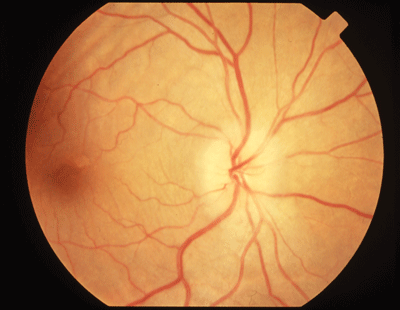 |
|
Optic neuritis more commonly occurs due to a demyelinating event than a systemic inflammatory process or infection, such as Lyme disease, and is a hallmark sign of multiple sclerosisthe most common cause of nerve fiber demyelination. |
The Optic Neuritis Treatment Trial (ONTT) demonstrated that the risk of CDMS with one or more lesions seen on MRI was 56% at 10 years and only 22% in patients who had normal MRI scans.40 The ONTT showed that patients with two or more lesions on MRI who were treated with high-dose IV methylprednisone had a 50% less chance of developing CDMS vs. those treated with placebo or oral prednisone.40,41 In fact, oral prednisone alone was found to increase the risk of recurrent optic neuritis in the same or fellow eye.40-41 However, subsequent follow-up of the ONTT results found the use of IV steroids offered only short-term reduction; by three years post-treatment, the protective benefits ceased.42
Alternatively, the Controlled High-Risk Subjects Avonex Multiple Sclerosis Prevention Study (CHAMPS) demonstrated that the risk of developing CDMS was further reduced from 50% in the placebo group to 35% in the treatment group when the patient was treated with weekly injections of Avonex (interferon-beta1a, Biogen Idec), an immunomodulating agent. Also, there was a reduction in the number of lesions detected by MRI at 18 months.43-45
Visual prognosis is good in optic neuritis, and many patients recover to 20/20 or near 20/20 acuity.37-39 However, most patients experience decreased visual function due to permanent changes in contrast sensitivity, color saturation and visual fields. Visual recovery is typically rapid, beginning within two weeks and continuing up to one year, regardless of treatment.39
So, what was the outcome of our wrongly diagnosed glaucoma patient? His glaucoma medications were discontinued, and he has been followed regularly with pressure checks, dilated examinations and field perimetry. His last IOP readings were 15mm Hg O.U. without Travatan, and his visual acuity and fields have remained stable.
While this patients correct diagnosis of NAAION may not have saved his vision, it could have saved him years of costly medication. As an aside, I have found that some practitioners support the use of ocular anti-hypertensives for prophylaxis in discs at risk. This patient, however, showed minimal response to the medications used.
Dr. McCann is a full-time faculty member at the Illinois College of Optometry in Chicago.
1. Chapter 4. Congenital and Acquired Anomalies of the Optic Nerve Head. In: Alexander LJ, ed. Primary Care of the Posterior Segment. 3rd ed. New York: McGraw Hill; 2002:257, 290-5.
2. Mathews MK. Nonarteritic anterior ischemic optic neuropathy. Curr Opin Ophthalmol 2005 Dec;16(6):341-5.
3. Arnold AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuro-Ophthalmol 2003 Jun;23(2):157-63.
4. Gray LG. NAAION: diagnosing in the negative. Rev Optom 2000 June 15; 137(6):83-98.
5. Characteristics of patients with nonarteritic ischemic optic neuropathy eligible for the Ischemic Optic Neuropathy Decompression Trial. Arch Ophthalmol 1996 Nov;114(11):1366-74.
6. Johnson LN, Arnold AC. Incidence of nonarteritic and arteritic anterior ischemic optic neuropathy. Population-based study in the state of Missouri and Los Angeles County, California. J Neuroophthalmol 1994 Mar;14(1):38-44.
7. Hayreh SS, Zimmerman B. Visual field abnormalities in non-arteritic anterior ischemic optic neuropathy: their pattern and prevalence at initial examination. Arch Ophthalmol 2005 Nov;123(11):1554-62.
8. Chapter 7. Ischemic Optic Neuropathies. In: Miller NR, Newman NJ. Walsh & Hoyts Clinical Neuro-Ophthalmology: The Essentials. 5th ed. Baltimore: Williams & Wilkins, 1999:221-46.
9. Burde RM. Optic disk risk factors for nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 1993 Dec 15;116(6):759-64.
10. Purvin V, King R, Kawasaki A, Yer R. Anterior ischemic optic neuropathy in eyes with optic disc drusen. Arch Ophthalmol 2004 Jan;122(1):48-53.
11. McGwin G Jr, Vaphiades MS, Hall TA, Owsley C. Non-arteritic anterior ischaemic optic neuropathy and the treatment of erectile dysfunction. Br J Ophthalmol 2006 Feb;90(2):154-7.
12. Optic nerve decompression surgery for nonarteritic ischemic optic neuropathy (NAION) is not effective and may be harmful. The Ischemic Optic Neuropathy Decompression Trial Research Group. JAMA 1995 Feb 22;273(8):625-32.
13. Chapter 4. Congenital and Acquired Anomalies of the Optic Nerve Head). In: Alexander LJ, ed. Primary Care of the Posterior Segment. 3rd ed. New York: McGraw Hill; 2002:295-9.
14. Liu GT, Glaser JS, Schatz NJ, Smith JL. Visual morbidity in giant cell arteritis. Clinical characteristics and prognosis for vision. Ophthalmology 1994 Nov;101(11):1779-85.
15. Keltner JL. Giant cell arteritis. Signs and symptoms. Ophthalmology 1982 Oct;89(10):1101-10.
16. Bhatti MT, Tabandeh H. Giant cell arteritis: diagnosis and management. Curr Opin Ophthalmol 2001 Dec;12(6):393-9.
17. Danesh-Meyer H, Savino PJ, Gamble GG. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology 2005 Jun;112(6):1098-103.
18. Chapter 4. Congenital and Acquired Anomalies of the Optic Nerve Head. In: Alexander LJ, ed. Primary Care of the Posterior Segment. 3rd ed. New York: McGraw Hill; 2002:305-15.
19. Talks SJ, Mossa F, Elston JS. The contribution of macular changes to visual loss in benign intracranial hypertension. Eye 1998;12(Pt5):806-8.
20. Eggers HM, Sanders MD. Acquired optociliary shunt vessels in papilloedema. Br J Ophthalmol 1980 Apr;64(4):267-71.
21. Rowe FJ, Sarkies NJ. Assessment of visual function in idiopathic intracranial hypertension: a prospective study. Eye 1998;12(Pt 1):111-8.
22. Chapter 11,13. Papilledema Rhee DJ, Pyfer MF, eds. The Wills Eye Manual. Office and Emergency Room Diagnosis and Treatment of Eye Disease. 3rd ed. Philadelphia: Lippin-cott Williams & Wilkins; 1999:306-8.
23. Sugerman HJ, DeMaria EJ, Felton WL 3rd, et al. In-creased intra-abdominal pressure and cardiac filling pressures in obesity-associated pseudotumor cerebri. Neurology 1997 Aug;49(2):507-11.
24. Farb RI, Vanek I, Scott JN, et al. Idiopathic intracranial hypertension: the prevalence and morphology of sinovenous stenosis. Neurology 2003 May 13;60(9):1418-24.
25. Goh KY, Schatz NJ, Glaser JS. Optic nerve sheath fenestration for pseudotumor cerebri. J Neuroophthalmol 1997 Jun;17(2):86-91.
26. Burgett RA, Purvin VA, Kawasaki A. Lumboperitoneal shunting for pseudotumor cerebri. Neurology 1997 Sep;49(3):734-9.
27. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri). Ophthalmology 1998 Dec;105(12):2313-7.
28. Chapter 10. Toxic and Deficiency Optic Neuropathies. In: Miller NR, Newman NJ, eds. Walsh and Hoyt"s Clinical Neuro-ophthalmology. The Essentials. 3rd ed. Baltimore: Lippincott Williams & Wilkins; 1999:290-302.
29. Chapter 4. Congenital and Acquired Anomalies of the Optic Nerve Head. In: Alexander LJ, ed. Primary Care of the Posterior Segment. 3rd ed. New York: McGraw Hill; 2002: 303-5.
30. Sadun AA. Metabolic optic neuropathies. Semin Ophthalmol 2002 Mar;17(1):29-32.
31. Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am 2004 Sep;17(3):481-8.
32. Clare G, Colley S, Kennett R, Elston JS. Reversible optic neuropathy associated with low-dose methotrexate therapy. J Neuroopthalmol 2005 Jun;25(2):109-12.
33. Yu Wai Man CY, Chinnery PF, Griffiths PG. Optic neuropathiesimportance of spatial distribution of mitochondria as well as function. Med Hypotheses 2005; 65(6): 1038-42.
34. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004 Jan;23(1):53-89.
35. Chapter 11,18 Miscellaneous Optic Neuropathies. In: Rhee DJ, Pyfer MF, eds. The Wills Eye Manual. Office and Emergency Room Diagnosis and Treatment of Eye Disease. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1999: 317-8.
36. Zoumalan CI, Agarwal M, Sadun AA. Optical coherence tomography can measure axonal loss in patients with ethambutol-induced optic neuropathy. Graefes Arch Clin Exp Ophthalmol 2005 May;243(5):410-6.
37. Chapter 6. Optic Neuritis. In: Miller NR, Newman NJ ed. Walsh & Hoyts Clinical Neuro-Ophthalmology: The Essentials. 5th ed. Baltimore: Lippincott Williams & Wilkins; 1999: 196-220.
38. Rebolleda G, Muoz-Negrete FJ, Noval S, et al. New ways to look at an old problem. Surv Ophthalmol 2006 Mar-Apr;51(2):169-73.
39.The clinical profile of acute optic neuritis: Experience of the Optic Neuritis Treatment Trial. Optic Neuritis Study Group. Arch Ophthalmol 1991 Dec;109(12):1673-8.
40. Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol 2003 Jul;121(7):944-9.
41. Beck RW. The Optic Neuritis Treatment Trial: three-year follow-up results. Arch Ophthalmol 1995 Feb;113(2):136-7.
42. Beck RW, Cleary PA, Trobe JD, et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med 1993 Dec 9;329(24):1764-9.
43. CHAMPS Study Group. MRI predictors of early conversion to clinically definite MS in the CHAMPS placebo group. Neurology 2002 Oct 8;59(7):998-1005.
44. CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol 2001 Oct;132(4):463-71.
45. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Eng J Med 2000 Sep 28;343(13):898-904.
