Dry eye disease (DED) affects all parts of the lacrimal functional unit—including the cornea, conjunctiva, meibomian glands, lacrimal glands and the interconnecting innervation. As clinicians, we tend to detect dry eye by listening to patient complaints for DED-related symptoms and closely examining the cornea. This isn’t a misguided approach, considering the cornea is crucial to vision and is often the first structure compromised by DED.
This article delves into DED’s impact on the cornea, including what we know about the pathophysiology of the condition, the initiating insult and methods of progression.
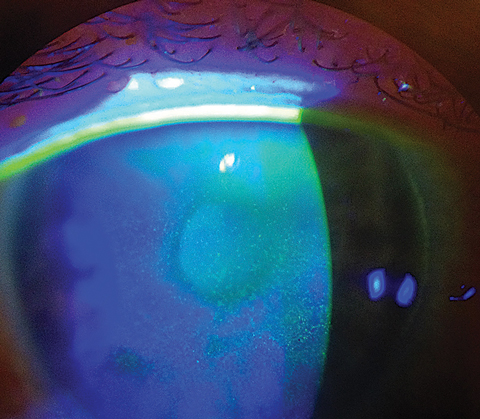 |
| This patient demonstrates diffuse punctate epitheliopathy, and shows decreased barrier function related to dry eye. |
Corneal Basics
The human cornea is an avascular structure comprised of several distinct layers.1 The outermost layer, the corneal epithelium, is composed of three stratified layers of cells: basal epithelium, wing cells and apical cells.1
Basal epithelium. This is in contact with the underlying basement membrane and adheres the epithelial complex to the structural portion of the cornea.1
Wing cells. These are in the middle section of the epithelium, and represent upwards migration of a portion of the basal epithelium in an intermediary state towards the apical cells.1
Apical cells. These are the outermost cells and are in direct contact with the tear film.1 The apical cells contain microvillae and microplicae, which extend upwards into the tear film as much as 0.5µm and are coated with a dense glycocalyx. Composed of several transmembrane mucins that form a mucoadhesive complex, the glycocalyx anchors the tear film to the apical epithelium and defends against bacteria.1
The cornea derives nutrients and oxygen from two different regions. The peripheral cornea derives its supply from the limbal region, which contains the vascular loops of the conjunctiva. The central anterior cornea—specifically the corneal epithelium—obtains most of its oxygen and nutrients from the tear film.
The epithelial surface is generated by the stem cell niches at the limbus, which produce transient amplifying cells.1 These migrate across Bowman’s membrane towards the central cornea, further from the limbus and blood supply. During normal cellular turnover, transient amplifying cell progenies will migrate across the cornea and differentiate into basal cells, and then into wing and apical epithelial cells.
As the most richly innervated tissue in the body, with receptors that number approximately 6000 sensory terminals per square millimeter, the cornea is designed to monitor and provide feedback for changes in the environment, including to the protective tear film.2,3 The majority of these corneal receptors are mechanoreceptors, which detect touch or pain; but approximately 10% to 15% respond to temperature and osmolarity changes, which may help detect evaporation and provide a means to regulate basal secretion.4
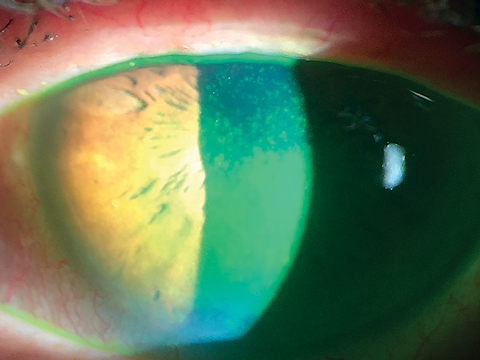 |
| Corneal staining shows advanced loss of epithelial barrier in severe dry eye with loss of the mucoadhesive layer and poor wettability of the tear film. Click image to enlarge. |
Healthy Tear Film
In a healthy eye, the tear film is stable and provides a sufficient refracting surface for good vision. It supports the ocular surface by providing oxygen and nutrients and protects it from dessicating environments, such as wind, and from toxins, microbes and particulate matter.
A healthy tear film contains a variety of constituent parts. It has a complex set and balanced number of electrolytes.5 It is pH neutral to slightly acidic.5 The tear film contains lysozyme and lacritin as defense mediators, as well as hundreds of proteins and proteolytic enzymes that support normal cellular function of the corneal epithelium and provide a protective barrier to the environment.5
Several glands on and around the ocular surface produce the constituent parts of the tear film, primarily the lacrimal and accessory glands, meibomian glands and conjunctival goblet cells.
Despite all of its important functions, the tear film in a healthy eye is extraordinarily thin, averaging between 2µm and 5.5µm in thickness over the cornea.6
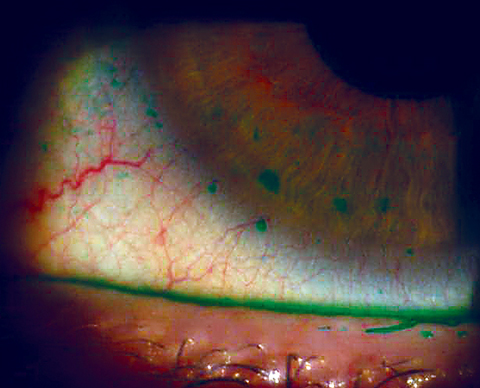 |
| In this patient, conjunctival epithelial compromise extends onto the cornea in macropunctate erosions, caused by dry eye. |
Homeostasis
This entire system is designed to monitor the environment and maintain homeostasis by compensating for alterations in temperature and humidity and protecting exposed tissue from bacteria and other offenders.
A normal eye maintains its homeostatic environment via this monitoring and compensatory actions. For example, the lacrimal functional unit’s ability to compensate and regulate tear production and volume is a hallmark of a healthy ocular surface system. Tearing is a compensatory reflex mechanism designed to increase or restore volume, to restore osmotic balance or to flush out toxins or irritants.
In the dry eye, patients suffer a loss of homeostasis, and the mechanisms normally designed to defend the eye—such as engagement of chronic inflammatory response— may turn against it and lead to DED progression. Returning to the example of tearing, it’s common for mild dry eye patients to experience tearing as a response to stress (e.g., when reading or exposed to a cold, drafty environment). In contrast, more severe dry eye patients, particularly those diagnosed with autoimmune conditions such as Sjögren’s syndrome, may have damaged glands and neurosensory systems due to chronic inflammation, preventing the ocular system from compensating to the stressors of increased evaporative demand.7
DED TestingSeveral tests can help you quantify the severity of dry eye, including vital dye staining of the cornea and conjunctiva, and point-of-care testing for the presence or degree of inflammatory involvement. Corneal and conjunctival staining is perhaps one of the most accessible and easily quantifiable methods of assessing damage to the ocular surface. Grading severity of corneal involvement is a key feature in determining DED severity. Several grading systems exist to help classify dry eye, such as the Oxford scheme, the Van Bjisterveld system and the NEI/Industry Workshop guidelines.19 Instillation of sodium fluorescein is a helpful diagnostic tool for grading damage to the ocular surface. Fluorescein staining will be present in cells with defective tight junctions, or a compromised glycocalyx.20 Remember, some mild background uptake of fluorescein dye is considered normal, so there is a weaker correlation between disease severity and stain uptake in mild forms of dry eye.21 Lissamine green, another vital dye well-tolerated by patients, will be taken up by epithelial cells with a damaged cell membrane and provides a means of assessing both the cornea and conjunctiva. While a number of testing options exist to provide a systematic means to assess the presence of staining on the cornea, no one particular method seems to have higher accuracy or better correlation to disease severity, so the testing method is entirely up to the clinician. |
Corneal Impact
The loss of homeostasis has a multitude of sequelae on the cornea, including visual complaints, increased risk for infection, keratitis and microerosions and, of course, inflammation.
Blurry vision. Because the cornea is dependent on the function of the thinnest portion of the tear film, blurry and inconsistent vision may be one of the earliest clues to a dry eye diagnosis. The Progression of Ocular Findings Study (PROOF), a five year, prospective, multicenter study, reveals one of the primary differences between individuals with mild DED and those without dry eye is how they perceive their quality of vision. In both groups, baseline distance best-corrected visual acuity (BCVA) was 20/20. However, 58.5% of those who had level two dry eye as defined by the International Task Force guidelines expressed moderate, severe or very severe dissatisfaction with their vision, vs. only 13.7% of those in the control group.8,9
Inflammation. Hyperosmolarity of the tear film is one of the key mechanisms that drives inflammation and the progression of DED. In addition, evaporative stress may be at the center of hyperosmolarity.
Studies examining osmolarity show that a difference in the osmolarity levels between the tear menisci and the precorneal tear film likely exists, with the precorneal tear film experiencing higher osmolarity.10 In patients with dry eye, this difference may be even greater. Mathematical models of osmolarity in the precorneal tear film show rapid increases due to evaporation and decreased tear break-up time. This may drive up the osmolarity of the precorneal tear film to levels that are quite high (e.g., 1,900mOsml) compared with normal (i.e., 300mOsml), especially where the tear film separates or collapses.11 In contrast, the highest readings in the tear meniscus measure well below 500mOsml.
On a cellular level, increased osmolarity triggers inflammation via cell signaling. Cell membrane receptors such as toll-like receptors (TLR) initiate the internal cell signaling processes, driven primarily by NF-kB and mitogen-activated protein kinase (MAPK) activation.12 These two cytoplasmic signaling pathways upregulate transcription of cytokines (primarily IL-1) and matrix metalloproteinases (MMPs). MMPs are substrate-specific proteolytic enzymes that regulate extracellular matrix deposition as well as its degradation.12
Keratitis. During periods of high evaporative stress, increases in tear film osmolarity trigger inflammation, beginning within the corneal apical cells. Increased tear film osmolarity alters the normal internal/external osmotic gradient, moving water and internal cell ionic content into the extracellular space. These result in cell volume losses, which cause gaps to form between adjacent epithelial cells, resulting in the appearance of “fine” superficial punctate keratitis (SPK) and increased penetration of fluorescein into the deeper layers of the corneal epithelium and then into the anterior stroma.
Recent measurements of SPK lesions seen in dry eye patients show that the mean diameters are smaller than a healthy epithelial cell. These are likely “deflated” or shrunken cells, which are in the process of apoptosis and have taken up dye.2
In more advanced forms of the condition, chronic epithelial compromise may also lead to mucin upregulation and formation of filamentary keratitis, where mucous adhesions cling to areas of chronic epithelial irregularity or compromise.
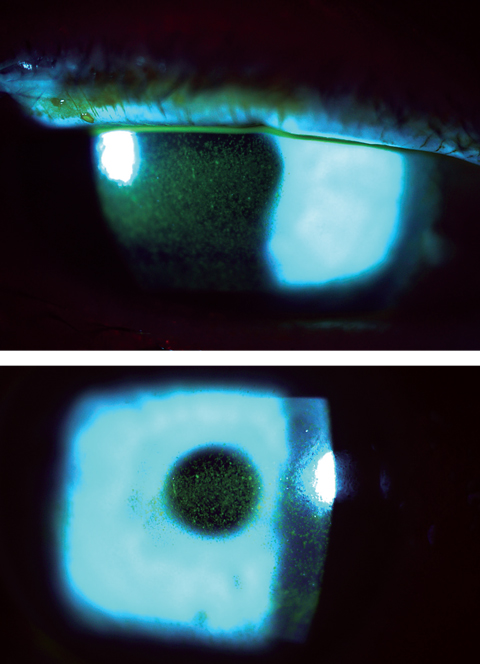 |
| This patient’s punctate epithelial keratopathy involves the central cornea in a case of Sjögren’s-related dry eye. Click image to enlarge. |
Infections. Dry eye continually arises as one of the top three risk factors for bacterial keratitis.13,14 Corneal integrity is necessary as an innate defense against bacteria. Chronic breaks in the surface of the cornea, in conjunction with decreases in lysozyme and lactoferrin, which provide a second defense against bacteria, predispose the dry eye to a higher risk of corneal infection. This becomes even more important for contact lens wearers, who are exposed to bacterial biofilms, as well as contact lens matrices that may become contaminated and populated over time by a number of bacteria, fungi and parasites.
On a cellular level, lymphocytic activation, recruitment and infiltration of tissues by T-lymphocytes, all hallmarks of DED, can ultimately decrease resistance to infection. Activation and recruitment of TH1/TH17 via the nuclear factor kappa beta (Nf-KB) signaling pathway is a critical part of the mechanism that drives the conversion from an acute response to chronic inflammation and seems to play a key role in disease progression.15,16 Infiltration of the lacrimal gland leads to damage and dysfunction of secretory acini. Ultimately, this alters the tear film composition.
Decreased corneal epithelial barrier molecules. While at least 30 known MMPs exist in the human body, MMP-3 and -9 appear particularly critical to maintaining the epithelial barrier of the cornea. For example, increased MMP-9 is correlated with increased corneal staining in dry eye patients, but is also increased in corneal infection, recurrent erosion and other pathologies where the corneal epithelial barrier is compromised.17 MMP-9 is normally found on the ocular surface in low amounts, below 41ng/mL in healthy corneas. Levels above 41ng/mL are associated with epithelial compromise. MMP-9 affects the junctions between epithelial cells by breaking down the molecule occludin, a critical component of the tight junctions.
As concentrations of pro-inflammatory molecules increase, compounds that maintain the corneal epithelium may decrease. The molecule extracellular matrix metalloproteinase inducer (EMMPRIN), a type-1 cornea epithelial cell receptor that may have an important role in the maintenance of the corneal epithelial barrier, increases in proportion with MMP-9 levels.18 Studies show an inverse relationship between EMMPRIN amounts and occludin in the cornea. Because occludin is critical to maintaining the tight junctions between adjacent epithelial cells, the overall barrier of the cornea is compromised.
Basal secretion, neurological changes and symptoms. In the presence of inflammation, damage to corneal nociceptors and associated neurologic pathways may decrease basal tear secretion as well as contribute to morphologic changes in nerve structure that may lead to sensations of pain or, conversely, to hypoesthesia and neurotrophia despite worsening disease.4,7
Mechanical epithelial desquamation. In a normal eye, the tear film provides a cushion and lubrication to help the lids move over the ocular surface with minimal friction. In a dry eye, however, both volume and content of the tear film is reduced, exposing the glycocalyx and apical epithelium to the frictional forces of the blink. This results in increased epithelial desquamation compared with normal eyes.
Dry eye disease has a number of negative effects on the ocular system, beginning with the breakdown of the homeostatic mechanisms designed to protect the health of the ocular surface. We can use the cornea as a window to assess the severity of DED, and once these systems are compromised, inflammatory insult to corneal and conjunctival tissues make it difficult to return to baseline homeostatic function. Processes that direct corneal wound healing and tight junction formation are compromised. Ongoing injury to the corneal barrier and reduced corneal integrity results in the appearance of reduced wettability due to glycocalyx disruption or loss, punctate keratopathy, microerosions, development of filaments and increased risk for infection.
By identifying issues early, we can make the task of restoring homeostasis of the ocular surface and preventing progression of the disease less daunting.
Dr. Hauswirth is an assistant professor in the Department of Ophthalmology at the University of Colorado School of Medicine, in Denver, CO.
1. Nishida T. Cornea. IN Krachmer JH, Mannis MJ, Holland EJ (eds) Cornea. 2nd Edition. Philadelphia, PA. Elsevier Mosby. 2005:3-26. |
