A 76-year-old female, accompanied by her husband, presented to the office as a new patient. She complained of decreasing vision O.U. Her medications included simvastatin, Cymbalta (duloxetine, Lilly), Synthroid (levothyroxine, Abbott), 81mg aspirin, and vitamins. In addition to her hypercholesterolemia and hyperthyroidism, the patient’s husband reported that she also had the early stages of dementia. After hearing this, I noticed mild tremors in the extremities and with her head and neck.
Best-corrected entering visual acuity was 20/30+ O.D. and 20/25- O.S. through hyperopic astigmatic correction. External examination was remarkable for moderate lid myokymia of the right eye, which was more notable in the lower lid. All other external findings were normal with no evidence of afferent pupil defect. Refraction was stable exhibiting negligible changes. Biomicroscopy was unremarkable O.U. Applanation tensions measured 17mm Hg O.D. and 15mm Hg O.S. at 10:15 a.m.
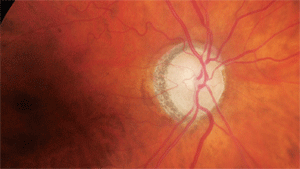
What can this glaucoma suspect’s optic nerve tell us about her neurodegeneration?
Through dilated pupils, her crystalline lenses were characterized by nuclear and cortical cataracts (O.D.>O.S.). Examination of her optic nerves revealed cup-to-disc ratios of 0.55 x 0.60 O.D. and 0.60 x 0.60 O.S. The neuroretinal rims were sloped toward the cup; though perfused, they lacked a distinct pink appearance. Both maculae were free of disease while her vascular evaluations demonstrated mild arteriolar narrowing. I asked the patient to return for a glaucoma evaluation.
Upon follow-up, her ocular health was unchanged. Her applanation tensions measured 15mm Hg O.D. and 16mm Hg O.S. at 9:50 a.m. Automated perimetry demonstrated normal visual fields. Pachymetry results measured 533µm O.D. and 546µm O.S. Gonioscopy demonstrated open angles visible to the scleral spur with normal trabecular pigmentation. Retinal topography demonstrated slightly thinned retinal nerve fiber layers.
Given these findings, I diagnosed the patient as a glaucoma suspect who did not yet require medical intervention. She was scheduled for a follow-up evaluation in six months.
Four months later, the patient presented on an urgent basis with complaints of pain and foreign body sensation in her left eye for the previous four days. Her husband reported that she had been rubbing the eye excessively for two weeks prior. Slit lamp examination showed entropion of the right lower lid with concurrent corneal eyelash contact and corneal disruption.
I fit and applied a bandage contact lens to the right eye, and prescribed a topical fluoroquinolone q.i.d. along with a topical non-steroidal anti-inflammatory (NSAID) q.i.d. for analgesia.
Upon follow-up three days later, she reported significant improvement in her discomfort and had decreased redness of the lids. Slit lamp examination demonstrated that her right cornea was re-epithelialized completely and only moderate episcleral injection in the left eye remained. The anterior chamber was quiet. There was now a mild blepharospasm observed on the right side. The bandage lens was removed and the antibiotic was stopped. The patient was discharged on a regimen of liberal use of artificial tears and instructions to return should the problem persist. The entropion was not constant at this follow-up, but the lid did have the tendency to occasionally become entropic.
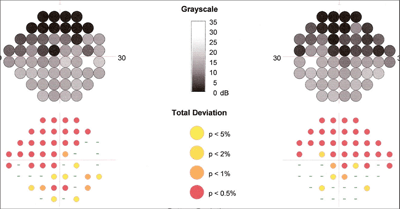
Heidelberg Edge Perimetry (HEP) revealed scattered paracentral defects that were consistent with early glaucomatous field loss.
Approximately three months later, the patient presented again with similar symptoms. On this visit, like the last visit, the blepharospasm of the right lower eyelid had produced entropion and corneal epithelial defects. Corneal rehabilitation was accomplished using the same technique as before, but it was apparent that the spastic entropic right lower lid was going to cause the epithelial defects again. So, the patient was injected with eight units of Botox (onabotulinumtoxinA, Allergan) in the lower lid as well as an additional 15 units in the upper lid.
Interestingly, her overall motor skills and fine motions had deteriorated significantly to the point where she was continuously moving her arms, legs, trunk, neck and head in a pronounced manner. Her husband relayed that she had been to a neurologist for her Parkinson’s disease and similar processes.
She returned for periodic follow-up visits during the next several months with no complaints of irritation to the right eye, and no blepharospasm or entropion. Applanation tensions remained in the mid-teens, and Heidelberg Edge Perimetry (HEP) revealed scattered paracentral defects that were consistent with early glaucomatous field loss O.U. The optic nerves appeared unchanged.
Glaucoma and Alzheimer’s
Is there possibly a link between this patient’s early dementia, loss of motor skills, and optic nerve appearance? Several years ago, widespread scientific news reported that “having an eye examination” could provide an early diagnosis of Alzheimer’s disease (AD). At that time, several novel studies looked at optic nerve characteristics in patients who were diagnosed with middle- to late-stage AD. Researchers were looking at various models that might link the diagnosis of AD to something else that would facilitate a much earlier diagnosis. The norm at the time was to wait until a constellation of symptoms developed and then make the diagnosis. As with any disease, early diagnosis would lead to early intervention.
The optic nerve became the focus of research because of its unique ability to be visualized (compared to other, less accessible central nervous system structures). A 1986 study compared the optic nerves and retinae of postmortem patients diagnosed with AD to those without AD.1 The researchers found that there was a significant decrease in the number of retinal ganglion cells in the optic nerves and retinal nerve fiber layers of patients with AD.
In 1989, researchers looked at the physical characteristics of the retinal nerve fiber layer (RNFL) and ganglion cells in humans with AD.2 Though this study also looked at postmortem tissues, fundamental differences were seen histologically in the RNFL and ganglion cells of patients with AD—namely, a loss of the glial cell support structure, as well as cytoplasmic changes in the retinal ganglion cells. The authors then suggested that optic nerve evaluation should become a standard part of the analysis of patients with AD.
Interestingly, a postmortem study of optic nerves in patients with AD, published in 1990, found that the predominant type of retinal ganglion cell lost in patients with AD was the M retinal ganglion cell.3 These cells are the large caliber ganglion cells believed to be susceptible to early glaucomatous damage and are preferentially evaluated by selective perimetry, such as the HEP (flicker defined form) strategy.
AD, Dementia and Parkinson’s
Both AD and Parkinson’s disease (PD) are currently classified as neurodegenerative diseases—diseases that manifest a myriad of symptoms because of the degeneration of the neurons and associated support structures on the central nervous system (CNS) and peripheral nervous system (PNS).
By definition, AD is a form of dementia characterized by the loss of several important mental functions.4 It is the most common manifestation of dementia.4 Dementia includes memory impairment as well as increased language difficulty, loss of motor skills and the inability to identify objects.
As AD progresses, the patient suffers an increase in mood swings, confusion, long-term memory loss, a progressive loss of bodily functions and ultimately death.4
Parkinson’s disease is also characterized by progressive degeneration of neurons in the CNS. Patients typically have trouble with locomotion in the early stages, including difficulty walking and fine use of the hands, as well as shaking and slowed movements.
As the disease progresses, cognitive ability becomes impaired. Most patients with PD have an idiopathic etiology, but a small number develop it because of genetic factors and mutations in the genes for alpha-synuclein.5
The main site of pathology in PD is dopaminergic innervated centers in the CNS, which is characterized by cell (neuronal) death, with the substantia nigra being primarily involved.
AD, on the other hand, is probably multifactorial in origin. One thought is that AD is brought on by reduced synthesis of acetylcholine.6 In fact, most current therapies for AD are geared toward mitigating this reduction in acetylcholine. Excessive amyloid buildup in the CNS is another postulated etiology.7-9 Oxidative stress may also play a role.10,11
The Optic Nerve, RNFL, AD and PD
Current research is looking at identifying the exact mechanisms for the beginning of the process of neuronal cell loss. Cerebral vascular endothelium breakdown and subsequent breakdown of the blood brain barrier is believed to play a role in not only the development of AD and PD, but also multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) and traumatic brain injury.12,13
As clinicians, what can we look for? One 2003 study from Italy showed that patients with pressure-dependent glaucoma, ocular hypertension and AD all show similar optic nerve characteristics—namely, a thinning of the RNFL, increased cupping and abnormal pattern electroretinogram (ERG) recordings.14
More recently, the use of optical coherence tomography (OCT) in the field of neurology has been gaining wider interest. Investigators have found that RNFL thickness, as measured by OCT, is diminished in patients with MS, AD and PD.15 While we cannot establish a definite cause-and-effect relationship between ocular hypertension, glaucoma and neurodegenerative diseases, there clearly is an overlap in the clinical findings in the eye.
One interesting hypothesis: Cerebrospinal fluid pressure may play a role in both AD and glaucoma as a correlation between low cerebrospinal fluid pressure is common in patients with glaucoma and AD.16
As discussed earlier, the protein alpha-synuclein has been implicated in the development of PD.5 Recently, gamma-synuclein has been found to be elevated in the optic nerves in patients with glaucoma.17 Gamma-synuclein is very similar to alpha-synuclein, and their respective aberrant forms are found in elevated levels in glaucoma and PD respectively.
Axons and Vision
When we look at glaucoma patients in clinic, the first thing that comes to mind is the diseased optic nerve. But if we view glaucoma as a neurodegenerative disease, we can see the parallels between it and AD and PD. We know that, in glaucoma, retinal ganglions die and the loss of these axons is seen clinically at the optic nerve. Axonal transport is disrupted, and the entire nerve fiber is lost, from the retinal ganglion cell layer to the lateral geniculate nucleus.
In a mouse model where elevated IOP was induced, researchers suggest that vision loss begins with axonal loss in the mid brain and that the axonal damage progresses anteriorly to eventually involve the portion of the optic nerve that is visible fundoscopically.18 Similar findings are seen in primates with elevated intraocular pressure.19,20
In our patient’s case, she exhibited progressive difficulty with motor skills, was in the early stages of Alzheimer’s, and was a glaucoma suspect based on the appearance of her optic nerves. Is there a link? Quite possibly. The most intriguing findings were the field defects obtained with the HEP perimeter, as those defects arise from damage to the M-cells of the retinal ganglion cells. Given the similarities of structural characteristics between patients with glaucoma and AD and PD, and the possibility that early neurodegenerative process preferentially damages the M-cells, was her HEP field defect related to just glaucoma, or perhaps to the other neurodegenerative processes as well?
Dr. Fanelli is in private practice in Wilmington, N.C., writes Review of Optometry’s “Glaucoma Grand Rounds” column, and lectures on glaucoma and other clinical topics.
1. Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic-nerve degeneration in Alzheimer’s disease. N Engl J Med. 1986 Aug 21;315(8):485-7.
2. Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Res. 1989 Nov 6;501(2):364-72.
3. Sadun AA, Bassi CJ. Optic nerve damage in Alzheimer’s disease. Ophthalmology. 1990 Jan;97(1):9-17.
4. Dementia: Hope Through Research. National Institute of Neurological Disorders and Stroke website. Available at: www.ninds.nih.gov/disorders/dementias/detail_dementia.htm. Accessed June 13, 2011.
5. Cullen V, Sardi SP, Ng J, et al. Acid ß-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol. 2011 Jun;69(6):940-53.
6. Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999 Feb;66(2):137-47.
7. Shen ZX. Brain cholinesterases: II. The molecular and cellular basis of Alzheimer’s disease. Med Hypotheses. 2004;63(2):308-21.
8. Wenk GL. Neuropathologic changes in Alzheimer’s disease. J Clin Psychiatry. 2003;64 Suppl 9:7-10.
9. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991 Oct;12(10):383-8.
10. Su B, Wang X, Nunomura A, et al. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res. 2008 Dec;5(6):525-32.
11. Kastenholz B, Garfin DE, Horst J, Nagel KA. Plant metal chaperones: a novel perspective in dementia therapy. Amyloid. 2009;16(2):81-3.
12. Grammas P, Martinez J, Miller B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev Mol Med. 2011 Jun 10;13:e19.
13. Guo L, Duggan J, Cordeiro MF. Alzheimer’s disease and retinal neurodegeneration. Curr Alzheimer Res. 2010 Feb;7(1):3-14.
14. Parisi V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer’s disease. Semin Ophthalmol. 2003 Jun;18(2):50-7.
15. Jindahra P, Hedges TR, Mendoza-Santiesteban CE, Plant GT. Optical coherence tomography of the retina: applications in neurology. Curr Opin Neurol. 2010 Feb;23(1):16-23.
16. Wostyn P, Audenaert K, De Deyn PP. Alzheimer’s disease and glaucoma: is there a causal relationship? Br J Ophthalmol. 2009 Dec;93(12):1557-9.
17. Nguyen JV, Soto I, Kim KY, et al. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A. 2011 Jan 18;108(3):1176-81.
18. Crish SD, Sappington RM, Inman DM, et al. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010 Mar 16;107(11):5196-201.
19. Gupta N, Ly T, Zhang Q, et al. Chronic ocular hypertension induces dendrite pathology in the lateral geniculate nucleus of the brain. Exp Eye Res. 2007 Jan;84(1):176-84.
20. Gupta N, Ang LC, de Tilly LN, et al. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br J Ophthalmol. 2006 Jun;90(6):674-8.
