In addition to treating the uveitis, the optometrist often must evaluate the patient for underlying etiologies and comanage with internal medicine, rheumatology and infectious disease physicians.
This article reviews the common symptoms and clinical findings, with a goal of helping the clinician determine the correct diagnosis and etiology, and providing the most appropriate treatment and care. Classifications Inflammation of the uvea (iris, ciliary body and choroid) is termed uveitis; however, there is a more precise classification system that depends on the location of the structure(s) involved. In addition, uveitis is often classified based on the onset and duration.
• Location. The International Uveitis Study Group (IUSG) has four classifications based on anatomic location of the primary source of inflammation: Anterior uveitis (anterior chamber), intermediate uveitis (vitreous), posterior uveitis (retina and choroid).1 The fourth classification, panuveitis, is used when there is no predominant site of inflammation, with involvement of the anterior chamber, vitreous, retina and/or choroid.2
• Onset. The Standardization of Uveitis Nomenclature (SUN) Working Group classifies onset as either “sudden,” which is characterized by pain, redness and photophobia, or “insidious,” where the eye is painless and white.2
• Duration. Duration is defined as either “acute,” where episodes have a sudden onset and limited duration, or “chronic,” with persistent relapses occurring less than three months after discontinuation of therapy.2 “Recurring” episodes are defined by repeating episodes that occur more than three months after discontinuation of therapy.2
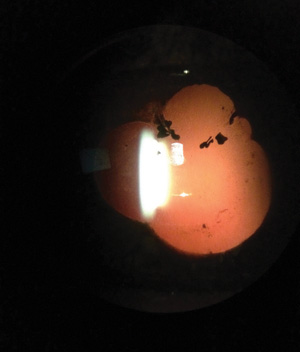 |
|
Unequal pupil dilation in a patient with posterior synechiae.
|
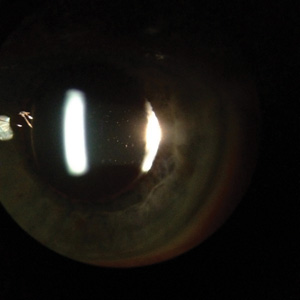
|
|
Cells and flare should be evaluated under high magnification after dark adap
tation.
|
Symptoms
• Acute pain. In acute cases of anterior uveitis, patients often present with pain (generally described as an ache in and around the eye), photophobia and redness. Pain and photophobia are a result of ciliary body inflammation and spasm, but can also be due to moderately elevated intraocular pressure .• Chronic pain. In cases of chronic anterior or intermediate uveitis, pain and redness are generally absent, although patients commonly note blurred vision and floaters.
• Blur/loss of vision. Depending on the severity of inflammation, presence of macular edema or media obstruction, vision may be unaffected, partially or significantly decreased.
Clinical Findings
A careful and detailed examination is required to properly differentiate and describe the findings of uveitis. These findings can vary based on the underlying etiology driving the inflammation. Let’s look at how each area can be involved.
• Conjunctiva. Perilimbal vessel engorgement of the conjunctival and episcleral vasculature (ciliary flush) is a characteristic finding of anterior uveitis. Diffuse injection can also be seen.
• Anterior chamber. Cells within the anterior chamber are a result of inflammatory cellular infiltration while flare is due to an influx of proteins. A grading system defined by the SUN Group helps quantify the amount of cells and flare seen on examination.2 (See “Standardized Grading Scales for Uveitis.") Evaluate cells and flare under high magnification following relative dark adaptation. The slit beam should be 1mm x 1mm at high intensity at a 45- to 60-degree angle.3
 Keratic precipitates (KPs) are cellular deposits of aggregated polymorphonuclear cells and lymphocytes located on the corneal endothelium.4 Classification of KPs is of clinical importance and may help narrow the differential diagnosis of any underlying cause. Non-granulomatous KPs are small, white precipitates on the posterior cornea while granulomatous KPs are larger and have a yellow or mutton-fat appearance.4 KPs generally deposit as an inverted triangle on the central to inferior cornea (Arlt’s triangle) due to aqueous convection currents.5 However, in patients with Fuchs’ heterochromic iridocyclitis, KPs are stellate and distributed over the entire corneal endothelium.4
Keratic precipitates (KPs) are cellular deposits of aggregated polymorphonuclear cells and lymphocytes located on the corneal endothelium.4 Classification of KPs is of clinical importance and may help narrow the differential diagnosis of any underlying cause. Non-granulomatous KPs are small, white precipitates on the posterior cornea while granulomatous KPs are larger and have a yellow or mutton-fat appearance.4 KPs generally deposit as an inverted triangle on the central to inferior cornea (Arlt’s triangle) due to aqueous convection currents.5 However, in patients with Fuchs’ heterochromic iridocyclitis, KPs are stellate and distributed over the entire corneal endothelium.4
Fibrin, generally associated with HLA-B27 uveitis, is due to a breakdown of the blood/aqueous barrier leading to a large amount of protein leakage.3 Increased intraocular pressure (IOP) can develop from accumulation of fibrin around the lens and iris.
Hypopyon formation results from an accumulation of white blood cells (WBCs) layering in the anterior chamber. It is most commonly seen with Behcet’s disease, HLA-B27 uveitis and herpetic uveitis.6 In addition to WBCs, pigmented cells from the iris or red blood cells from iris neovascularization or trauma may be noted on exam.
• Iris and pupil. Persistent inflammation can cause scarring of the iris to anterior lens (posterior synechia) or adhesions of the iris to cornea (anterior synechia). Significant synechia formation can lead to elevations in IOP. In certain conditions that cause granulomatous uveitis, such as sarcoidosis, inflammatory nodules can be noted. Koeppe, Busacca and Berlin nodules are granulomas seen on the pupillary margin, iris and angle respectively.3
• Vitreous. According to the SUN Group, “intermediate uveitis” is when the primary source of inflammation is within the vitreous.2 Pars planitis, a subset of intermediate uveitis, describes snowbanking or snowball formation only in idiopathic cases; the term intermediate uveitis is used if there is an underlying infectious or autoimmune cause.2
• Posterior chamber. Posterior uveitis involves primary inflammation of the retina and/or choroid. Retinits and choroiditis can be focal, multifocal or diffuse.3 Vasculitis, macular edema and neovascularization are common complications seen as a result of posterior inflammation. The differential diagnoses of posterior uveitis are broad and include white dot syndromes, collagen vascular and infectious diseases.
• Intraocular pressure. Uveitis can either increase or decrease the intraocular pressure. Ciliary body inflammation results in a decrease of aqueous production leading to a decrease in IOP.7 Drops in IOP can be significant, although the risk of hypotony is less than 2%.8
Alternatively, an elevation in IOP can occur either from resistance to aqueous outflow by inflammatory cells and proteins, pupil block, inflamed trabecular meshwork (trabeculitis), or as a response to steroid therapy.7 Steroid responders generally develop an increase in IOP after two to six weeks of therapy, but it can occur at any point.7
Children are more susceptible to a steroid response than adults and generally develop increased IOP earlier on.9
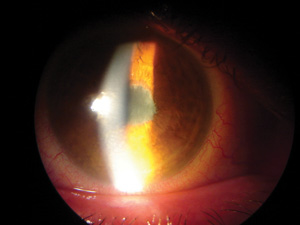 |
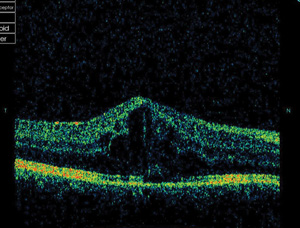 |
|
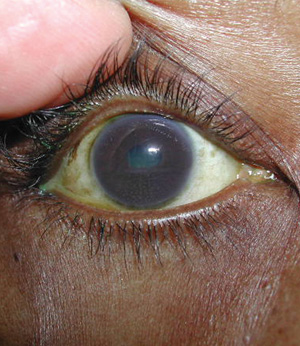
The iris is bound to the lens in this patient w
ith anterior uveitis
who waited one month before seeking treatment. |
In the same patient, an OCT scan shows macular edema that
is related to the uveitis. |
Etiologies of Uveitis
Because there are many causes of uveitis documented, we’ll discuss the more commonly associated conditions and their pertinent uveitic findings. Causes of uveitis can be broadly separated into non-infectious and infectious etiologies.Non-Infectious Etiologies
HLA-B27 Seronegative Spondyloarthropathies
The seronegative spondyloarthropathies are a group of inflammatory disorders with a negative rheumatoid factor and a strong relationship to the human leukocyte antigen (HLA)-B27. HLA-B27 is a major histocompatibility class 1 molecule.10 While it’s only found in about 8% to 10% of the general population, HLA-B27-associated uveitis accounts for 18% to 32% of anterior uveitis cases in the Western population, although the exact mechanism by which it causes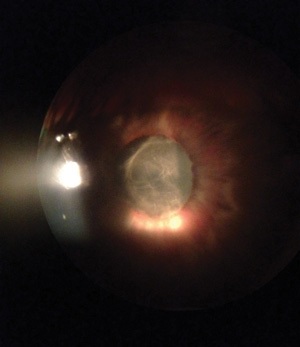 |
 |
|
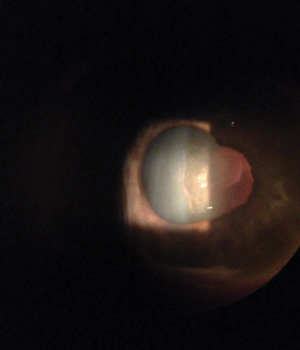
Fibrin mem
brane
formation often
presents in patients with HLA-B27 uveitis. |
The membrane start
s
to break
after
inserting a cotton pledget soaked in cyclopentolate, homatropine and phenyloephrine, . |
• Ankylosing spondylitis. This is a chronic inflammatory disorder that is the prototype of SpA. Ankylosing spondylitis (AS) is characterized by sacroiliitis, spinal inflammation and enthesitis (inflammation of the site where tendons and ligaments insert into the bone, commonly occurring at the heel near the Achilles tendon).12 The chronic inflammation found with AS leads to fibrosis and ossification mostly at the edges of inter-vertebral discs.13 There is a male predominance of 2:1, and symptoms can range from asymptomatic to debilitating.3
• Reactive arthritis syndrome. Formerly known as Reiter’s syndrome, reactive arthritis syndrome (RAS) has a classic triad of urethritis, polyarthritis and conjunctival inflammation. Non-granulomatous anterior uveitis is the second most common ocular finding after conjunctivitis, and is often acute and unilateral.3 Previous infection of the genitourinary or gastrointestinal tract in HLA-B27-predisposed patients is thought to play a role in the pathophysiology of RAS.14 It occurs most commonly in men between the ages of 20 to 35.14
• Psoriatic arthritis. This as an inflammatory joint condition associated with skin psoriasis.15 Commonly, dermatologic changes are noted decades before arthritis, and nail pitting is frequently seen.15,16 Peak incidence is between 40 to 50 years, but can occur at any age, with a slight male predominance.
• Inflammatory bowel disease. This term encompasses a variety of different conditions, with the main two types being Crohn’s disease (CD) and ulcerative colitis (UC).17 Both of these conditions are characterized by chronic inflammation of the gastrointestinal tract. CD affects the entire intestinal tract (mouth to anus) with a patch-like pattern of inflammation, while UC affects mainly the large intestine as a continuous area of inflammation.For all types of HLA-B27 spondyloarthropathies, recommended testing includes HLA-B27, rheumatoid factor (RF), and imaging of the spine and sacroiliac joint.
Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA)—also known as juvenile rheumatoid arthritis and juvenile chronic arthritis—is the most common cause of arthritis in children under the age of 16.3 The subsets of JIA are divided by the mode of presentation and the level of joint inflammation within the first six weeks:• Oligoarticular onset (previously named pauciarticular) involving four or fewer joints, categorized as either persistent or extended. This is the most common form of JIA, affecting 60% of patients.18
• Polyarticular onset involves five or more joints. This subset is further divided into RF positive or RF negative groups. Patients in the RF positive group rarely develop uveitis.3
• Systemic onset (also known as Still’s disease) presents with fever and rash, lymphadenopathy, heptomegaly or splenomegaly.14 Of the subsets of JIA, the oligoarticular form is the most likely to be associated with the development of uveitis.3 The clinical presentation of JIA-associated uveitis is typically a chronic bilateral nongranulomatous anterior uveitis, more frequently affecting girls. Due to the chronic nature of inflammation, patients may also develop cataracts or band keratopathy.3 Laboratory testing of suspect patients should include RF and anti-nuclear antibody (ANA).
Sarcoidosis
 |
|
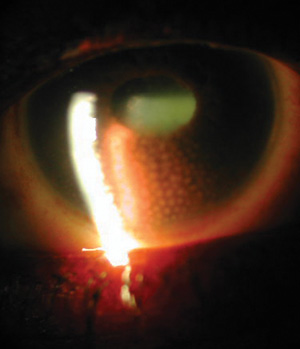
Note the granulomatus keratic precipitates
in this patient with sarcoidosis-related uveitis. |
 |
|
Characteristic "mutton-fat" keratic precipitates,
deposited as an inverted trian
gle on the infer
ior
cornea (Arlt's triangle).
|
Recommended lab tests include angiotension converting enzyme (ACE), serum lysozyme and chest radiography.
Systemic Lupus Erythematous
Systemic lupus erythematous (SLE) is a multi-organ connective tissue disorder that occurs more frequently in women.20 A type III hypersensitivity reaction, SLE is a disease in which B cells produce autoantibodies directed toward the DNA, cytoplasm and cell membrane.21 This results in inflammation, vasculitis, immune complex deposition, vasculopathy and end-organ damage.20Anterior uveitis from SLE seldom occurs in isolation and is more commonly associated with scleritis or posterior uveitis.21 Lupus retinopathy and choroidopathy indicate systemic disease activity and can present with retinal vasculitis, neovascularization and serous exudation.22 Antibody testing in suspected cases of SLE can include ANA, anti-SM, anti-dsDNA, anti-SSa/anti-SSb, anti-RNP and anticardiolipin (ACA).
Behcet’s Disease
Behcet’s disease (BD) is a multi-organ and multisystem chronic, relapsing, occlusive vasculitis. Although the etiology is unknown, BD has been associated with HLA-B51.3 BD is characterized by its triad of oral ulcers, genital ulcers and uveitis. Diagnosis is based mostly on clinical findings.BD is associated with a nongranulomatous anterior and/or posterior uveitis, KPs, posterior synechiae and normal to low IOP.23 Up to 25% of cases present with hypopyon, which typically indicates worse visual prognosis.23Variable amounts of vitritis, necrotizing vasculitis and cystoid macular edema can be found posteriorly.23 There is no specific laboratory test for BD, but testing can include HLA-B51 and a skin pathergy test.
Vogt-Koyanagi-Harada Disease
Vogt-Koyanagi-Harada (VKH) disease is a multisystem autoimmune disorder principally affecting pigmented tissues in the ocular, auditory, integumentary and central nervous systems.24 The pathogenesis of VKH is thought to be related to an aberrant T cell-mediated immune response directed against self-antigens found on melanocytes.24 VKH affects mainly darkly pigmented populations, including East and Southeastern Asians, Asian Indians, Middle Easterners, Hispanics and Native Americans; people of European and African descent are rarely affected.24VKH presents in four stages: prodromal, acute uveitic, convalescent and chronic recurrent.3,24 The prodromal phase is marked by flu-like symptoms. The acute uveitic stage presents as a diffuse, bilateral, granulomatous anterior uveitis.24 There may be some vitritis and choroiditis along with multiple, serous retinal detachments.3,24 Mutton-fat KPs, iris nodules and increased IOP can also be present.3 In the convalescent stage, depigmentation occurs, affecting the skin (vitiligo), eyelashes (poliosis) and choroid, giving the fundus a “sunset glow” appearance.25
The relapse of uveitis is what constitutes the chronic recurrent stage.HLA associations have been reported with VKH, but they are neither diagnostic nor required.
Fuchs’ Heterochromic Iridocyclitis
Fuchs’ heterochromic iridocyclitis (FHI) is a chronic, low-grade, unilateral nongranulomatous anterior uveitis that accounts for 2% to 3% of all uveitis cases.26 Patients are usually asymptomatic. Signs are typically mild, with little to no conjunctival injection.26 Despite persistent cells and flare, synechiae rarely occurs.26 With FHI, KPs have a characteristic diffuse, stellate appearance.4,26Heterochromia is a key diagnostic finding in FHI—the lighter iris (which can have a “moth-eaten appearance”) represents the involved eye.26 This can vary based on iris pigmentation and level of stromal atrophy.26 Reversed heterochromia is also possible, especially in lighter-eyed patients; in such a case, the darker iris represents the eye with inflammation due to stromal atrophy exposing large areas of iris-pigmented epithelium.26 Diagnosis of FHI is largely clinical and no routine testing is needed. Treatment of FHI is directed toward bouts of increased inflammation. Although topical corticosteroids lessen inflammatory findings, they do not eliminate them.14 Common sequelae of FHI are cataracts and glaucoma, the latter of which is often difficult to manage.3,14
Infectious Etiologies
Herpes Virus
• Herpes simplex. Herpes simplex virus (HSV), a member of the herpesvirus family, is acquired via direct contact of an active lesion. In most infected individuals, the virus remains latent in the neural ganglia. Once activated, HSV causes painful vesicular lesions in the corresponding area that the ganglia supplies. Virus reactivation can be induced by several factors such as stress, illness or sunlight exposure. Malaise and fever may accompany an active infection and skin lesions generally last for one to two weeks.HSV anterior uveitis is most commonly unilateral, associated with diffuse endothelial KPs and causes elevated IOP. So, unilateral uveitis with significantly elevated IOP generally indicates herpetic uveitis. Treatment of HSV anterior uveitis requires topical and sometimes oral corticosteroids, oral antiviral medicines and, when indicated, anti-glaucoma medications for elevated IOP. HSV IgG and IgM antibodies can be tested in cases when the clinical course is questionable; however, negative serology doesn’t exclude the diagnosis, as sensitivity to serologic testing is poor.
• Herpes zoster. Varicella zoster virus (chicken pox) causes an acute infection generally occurring in childhood. Similar to HSV, the herpes zoster virus remains latent in the neural ganglia until reactivated. A prodromal phase consisting of general malaise, fever and paraesthesia can occur before skin lesions appear. An eruptive phase of painful vesicular lesions, following the affected dermatome, generally lasts one to two weeks.
Zoster-related uveitis can be acute in conjunction with the eruptive phase or persist chronically. The uveitis always occurs on the same side as the affected dermatome. Less common findings are retinitis, progressive outer retinal necrosis or multifocal choroiditis. Diagnosis of herpes zoster virus is based largely on clinical findings and laboratory testing is not required.
Acute herpes zoster virus is treated with oral antivirals, and anterior uveitis is managed with corticosteroids.
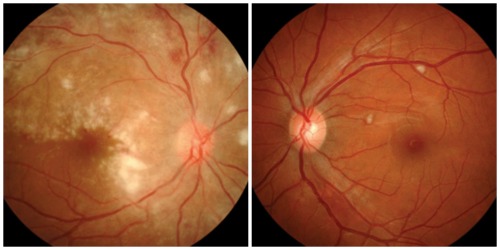 |
|
HIV positive patient who presented with bilateral retinitis
, OD>OS. The patient presented with dull eye
pain, decreased vision and floaters. Exam revealed cotton-wool spots, retinal hemmorrhages, exudate and perivascular sheathing. A moderate vi tritis, OD>OS, was noted on exam as well. |
Syphilis
Acquired syphilis is a sexually transmitted disease caused by the spirochete Treponema pallidum. It can infect multiple organ systems and has been called the “great masquerader” because its appearance is similar to many other diseases.27 The phases of syphilis infection are:• Primary. T. pallidum replicates at the site of initial inoculation and induces a painless chancre that occurs three to six weeks after infection.28
• Secondary. This occurs four to 10 weeks after primary infection and its most common clinical manifestation is a disseminated maculopapular rash.28 Malaise, fever, headache, hepatitis and meningitis can also occur.28
• Latent. The latent phase occurs as the clinical findings of secondary syphilis resolve and most patients become asymptomatic. Recurrences of secondary syphilis are common.
• Tertiary. Approximately one-third of patients with untreated latent syphilis develop tertiary syphilis.3 Although rarely seen, manifestations of tertiary syphilis are gummas or granulomatous lesions affecting multiple organs, aortic aneurysm and syphilitic meningitis.28
Syphilitic uveitis can be unilateral or bilateral, granulomatous or nongranulomatous, and affect the anterior, intermediate or posterior segment.3 Vascularized papules (iris papulos) or red nodules (iris nodosa) can be seen with iridocyclitis.3 Retinitis, chorioretinitis and vitritis can also occur. Fluorescent treponemal antibody-absorption (FTA-ABS) has a high sensitivity for diagnosis of syphilis.
Tuberculosis
Tuberculosis (TB) is a disease caused by the acid-fast Mycobacterium tuberculosis. Commonly affecting the lungs, TB is responsible for 0.6% of cases of uveitis in the US.29The clinical manifestations of intraocular TB include acute anterior uveitis, chronic granulomatous anterior uveitis, intermediate uveitis, vitritis or endophthalmitis.29 Granulomas can be noted on the iris, angle or choroid.Testing for TB can include purified protein derivative (PPD), and chest radiography. An anergy panel is used as an adjunct to PPD in patients who are immunocompromised.
Lyme Disease
Lyme disease (LD) is caused by the spirochete Borrelia burgdorferi and is transmitted via tick bites. The Centers for Disease Control and Prevention (CDC) estimate that 300,000 people are infected with LD each year, mostly in the northeastern and upper midwestern states.30 There are three stages of LD:• Stage 1 is early-localized disease presenting with classic bull’s-eye rash and fever.31
• Stage 2 occurs as the infection disseminates and patients can develop cardiac, neurologic and arthritic manifestations.31
• Stage 3 commonly manifests as Lyme arthritis, along with neuropsychiatric dysfunction.31
Uveitis secondary to LD is most commonly reported with stage 2 and 3 disease and can present as all forms of uveitis.3 When LD is suspected, enzyme-linked immunosorbent assay (ELISA) is used to identify B. burgdorferi antibodies. If the ELISA is positive, a Western blot test is performed to confirm the diagnosis .
Toxoplasmosis
Toxoplasmosis is caused by the parasite Toxoplasma gondii, which is commonly contracted from eating undercooked meat or from exposure to cats or cat feces.14,32Toxoplasmosis presents as a unilateral retinochoroiditis with creamy-white retinal necrosis and dense overlying vitritis giving the appearance of “headlights in a fog.”14,32 New retinal lesions are typically found adjacent to areas of old lesions. Additionally, perivasculitis, macular edema, subretinal neovascularization and mild anterior chamber reaction can occur.14,32
Suspected cases of toxoplasmosis can be confirmed with toxoplasma IgG and IgM serology.
Histoplasmosis
Histoplasmosis is fungal infection due to contact with spores of Histoplasma capsulatum with a high incidence found along the Ohio and Mississippi river valleys.33 Ocular histoplasmosis syndrome (OHS) is a chorioretinitis and presents with the triad of peripapillary atrophy, peripheral chorioretinal atrophy (histo-spots) and maculopathy.14,33 OHS is limited to the posterior segment and the absence of vitritis is key for diagnosis.14,33 Laboratory testing is not required but can include chest radiograph or histoplasma skin testing.14
Treatment
Treatment of uveitis largely depends on the severity of inflammation. Goals of treatment should be aimed at reducing the ocular inflammation and managing any associated complications.
• Anti-inflammatories. Corticosteroids are the mainstay treatment of uveitis. The frequency of dosing is individualized based on the amount and location of inflammation. The two major topical corticosteroids for uveitis are Pred Forte (prednisolone acetate 1%, Allergan) and Durezol (difluprednate 0.05%, Alcon). Durezol is indicated for treatment of inflammation and pain associated with ocular surgery as well as treatment of endogenous anterior uveitis. It has been shown to be as effective (dosed QID) as Pred Forte 1% (dosed eight times/day) for the treatment of inflammation and pain associated with anterior uveitis.34 The advantage of difluprednate (an emulsion) over prednisolone (a suspension) is not only the reduced frequency of administration, but also the elimination of shaking before use.
Other topical corticosteroids, such as Lotemax (loteprednol 0.5%, Bausch + Lomb), carry the benefit of having less IOP increase, but are not quite as effective as prednisolone in treating anterior uveitis.35
Oral steroids can be used to complement topical therapy or if there is an underlying systemic cause that requires treatment. Prednisone is used most commonly and the dosage is individualized based on the amount of inflammation, but generally 1mg/kg/day. In order to minimize the gastrointestinal effects of prednisone, proton pump inhibitors, such as omeprazole, or H2-blockers, such as ranitidine, are used in conjunction. Vitamin D and calcium supplementation is recommended with longer use to prevent osteoporosis. Consult with the patient’s internist when oral administration of prednisone may exacerbate pre-existing conditions, such as diabetes, hypertension, chronic GERD, and in patients who are immunocompromised. Periocular corticosteroids using a sub-Tenon approach are indicated for uveitis unresponsive to topical treatment, intermediate or posterior inflammation, macular edema, or for patients who are non-compliant.3 Infectious etiologies that corticosteroids could exacerbate should be ruled out prior to administration.
 In cases unresponsive to topical, periocular and systemic steroids, or for control of macular edema, intravitreal steroid injections are used.36 Intravitreal triamcinolone has been shown to effectively reduce macular edema resulting from uveitis.37 The risk of sterile endophthalmitis may occur in 1% to 6% of patients receiving injections; however, the FDA recently approved Triesence (Alcon), a preservative-free triamcinolone.3 Fluocinolone acetonide (Retisert) is a sustained-release implant used for treating chronic non-infectious posterior uveitis. Although it provides good long-term control of inflammation, it has been associated with cataract formation and glaucoma.3
In patients with recalcitrant ocular inflammation, steroid intolerance or lack of response to steroid treatment, immunomodulating therapies (IMT) can provide a great benefit. They can be prescribed by the internist or rheumatologist to treat the underlying disease, but can also be used in idiopathic cases with chronic ocular inflammation. Additionally, IMT offers the benefit of corticosteroid sparing due to the side effects associated with chronic prednisone use. Methotrexate is a commonly used agent for treatment of chronic non-infectious uveitis. A retrospective case series of 160 patients showed methotrexate to adequately control uveitis in 76.2% of patients treated, with a steroid-sparing effect achieved in 56% of patients.38
In cases unresponsive to topical, periocular and systemic steroids, or for control of macular edema, intravitreal steroid injections are used.36 Intravitreal triamcinolone has been shown to effectively reduce macular edema resulting from uveitis.37 The risk of sterile endophthalmitis may occur in 1% to 6% of patients receiving injections; however, the FDA recently approved Triesence (Alcon), a preservative-free triamcinolone.3 Fluocinolone acetonide (Retisert) is a sustained-release implant used for treating chronic non-infectious posterior uveitis. Although it provides good long-term control of inflammation, it has been associated with cataract formation and glaucoma.3
In patients with recalcitrant ocular inflammation, steroid intolerance or lack of response to steroid treatment, immunomodulating therapies (IMT) can provide a great benefit. They can be prescribed by the internist or rheumatologist to treat the underlying disease, but can also be used in idiopathic cases with chronic ocular inflammation. Additionally, IMT offers the benefit of corticosteroid sparing due to the side effects associated with chronic prednisone use. Methotrexate is a commonly used agent for treatment of chronic non-infectious uveitis. A retrospective case series of 160 patients showed methotrexate to adequately control uveitis in 76.2% of patients treated, with a steroid-sparing effect achieved in 56% of patients.38
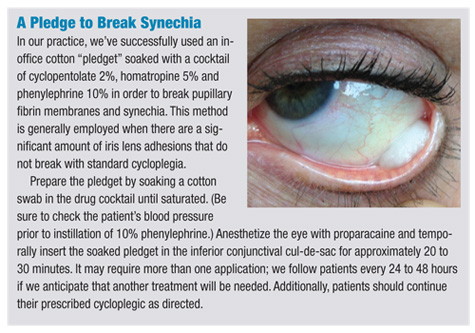
• Cycloplegics. Topical cycloplegics are used to treat pain associated with ciliary spasm, stabilize the blood-aqueous barrier and help prevent or break synechiae formation. Longer-acting cycloplegics such as homatropine, scopolamine and atropine are generally used. In cases of significant synechiae, a pledget can be instilled in the office. (See “A Pledge to Break Synechia .”)
• Glaucoma treatment. In the US, approximately 20% of patients with uveitis develop glaucoma.7 Uveitic glaucoma can be secondary to open or closed angle. Treatment of the inflammation with corticosteroids often controls the IOP, but the use of anti-glaucoma medicines is also very common. Topical prostaglandin analogs (PGAs), beta-blockers, carbonic-anhydrase inhibitors (CAIs) and alpha-adrenergic agonists all have been reported and used to control uveitic glaucoma. Typically, aqueous suppressants such as topical beta-blockers and CAIs are used in treating acute IOP spikes during active inflammation. When IOP cannot be controlled with topical medications, an oral CAI such as acetazolamide is used up to 1,000mg per day.7
When the ocular inflammation is quiescent, PGAs can be used to control intraocular pressure and are not associated with an increased risk of macular edema.39,40
When medical therapy is unsuccessful in controlling IOP, consider selective laser trabeculoplasty. Laser iridotomy can treat pupillary block from synechia or persistent fibrin membrane formation. Lastly, if all other therapies fail, trabeculectomy or valve implantation is required.7
Laboratory Testing
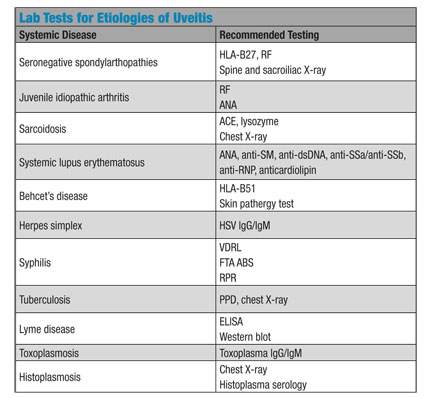 The most important steps prior to obtaining any laboratory tests are a careful and thorough history and clinical examination. Testing should be ordered based on signs and review of symptoms rather than taking a one-size-fits-all approach. Complete blood counts (CBC), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) can be included to indicate active systemic inflammation. Testing is not routinely done on asymptomatic individuals with uncomplicated initial episodes of mild non-granulomatous anterior uveitis. Indications for testing are: positive systemic history; recurrences, worsening or recalcitrant inflammation; bilaterality; granulomatous inflammation; and intermediate or posterior uveitis.
The most important steps prior to obtaining any laboratory tests are a careful and thorough history and clinical examination. Testing should be ordered based on signs and review of symptoms rather than taking a one-size-fits-all approach. Complete blood counts (CBC), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) can be included to indicate active systemic inflammation. Testing is not routinely done on asymptomatic individuals with uncomplicated initial episodes of mild non-granulomatous anterior uveitis. Indications for testing are: positive systemic history; recurrences, worsening or recalcitrant inflammation; bilaterality; granulomatous inflammation; and intermediate or posterior uveitis.
When attending to the patient with uveitis, be sure to first recognize the key characteristics of their presentation and then tailor the treatment and laboratory testing appropriately. Appropriate treatment and management of ocular inflammation helps prevent the complications of uveitis and preserves visual function. Optometrists can play an important role in not only the diagnosis and management of ocular findings with uveitis but also in uncovering the systemic causes as well.
Dr. Trottini is in practice at Outlook Eyecare, in Monroe Township, NJ. Dr. Tolud is in practice at South Jersey Eye Physicians, in Cream Ridge, NJ.
1. Bloch-Michel E, Nussenblatt RB. International Uveitis Study Group recommendations for the evaluation of intraocular inflammatory disease. Am J Ophthalmol. 1987 Feb 15;103(2):234-5.2. Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005 Sep;140(3):509-16. 3. Moorthy RS. 2011-2012 Basic and Clinical Science Course, Section 9: Intraocular Inflammation and Uveitis. San Francisco, CA: American Academy of Ophthalmology; 2011.4. MC Mocan, S Kadayifcilar, M Irkec. Keratic precipitate morphology in uveitic syndromes including Behçet’s disease as evaluated with in vivo confocal microscopy. Eye (Lond). 2009 May;23(5):1221-7.5. Van Gelder RN, Prasad AG. Review of Uveitis. Thorofare, NJ; Slack Inc; 2008: 33. 6. D’Alessandro LP, Forster DJ, Rao NA. Anterior uveitis and hypopyon. Trans Am Ophthalmol Soc. 1991;89:303-9.7. Siddique SS, Suelves AM, Baheti U, Foster CS. Glaucoma and uveitis. Surv Ophthalmol. 2013 Jan-Feb;58(1):1-10.8. Daniel E, Pistilli M, Pujari SS, et al. Risk of hypotony in noninfectious uveitis. Ophthalmology. 2012 Nov;119(11):2377-85.9. Yamashita T, Kodama Y, Tanaka M, et al. Steroid-induced glaucoma in children with acute lymphoblastic leukemia: a possible complication. J Glaucoma. 2010 Mar;19(3):188-90.10. Suhler EB, Martin TM, Rosenbaum JT. HLA-B27–associated uveitis: overview and current perspectives. Curr Opin Ophthalmol. 2003 Dec;14(6):378-83.11. Chang JH, McCluskey PJ, Wakefield D. Acute anterior uveitis and HLA-B27. Surv Ophthalmol. 2005 Jul-Aug;50(4):364-88. 12. Slobodin G, Rosner I, Rimar D, et al. Anklyosing spondylitis: field in progress. Isr Med Assoc J. 2012 Dec;14(12):763-7.13. Zambrano-Zaragoza JF, Agraz-Cibrian JM, González-Reyes C, et al. Anklyosing spondylitis: from cells to genes. Int J Inflam. 2013;2013:501653.14. Multack RF, Genge MR, Skorin L. Immune Disease. In: Onofrey B, Skorin L, Holdeman NR (eds). Ocular Therapeutics Handbook: A Clinical Manual, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:236-88.15. Goldenstein-Schainberg C, Favarato MH, Ranza R. Current and relevant concepts in psoriatic arthritis. Rev Bras Reumatol. 2012 Jan-Feb;52(1):98-106. 16. Gladman DD, Antoni C, Mease P, et al. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005 March; 64(Suppl 2): ii14–ii17. 17. Schoultz M, Atherton I, Hubbard G, Watson AJ. Assessment of causal link between psychological factors and symptom exacerbation in inflammatory bowel disease: a protocol for systematic review of prospective cohort studies. Syst Rev. 2013 Jan 23;2:8.18. Kahn P. Juvenile idiopathic arthritis :an update for the clinician. Bull NYU Hosp Jt Dis. 2012;70(3):152-66.19. Bonfioli AA, Orefice F. Sarcoidosis. Semin Ophthalmol. 2005 Jul-Sep;20(3):177-82.20. Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol. 2003 Jul;56(7):481-90.21. Sivaraj RR, Durrani OM, Denniston AK, et al. Ocular manifestations of systemic lupus erythematosus. Rheumatology (Oxford). 2007 Dec;46(12):1757-62.22. Nguyen QD, Uy HS, Akpek EK, et al. Choroidopathy of systemic lupus erythematosus. Lupus. 2000;9(4):288-98.23. Bonfioli AA, Orefice F. Behçet’s disease. Semin Ophthalmol. 2005 Jul-Sep;20(3):199-206.24. Damico FM, Kiss S, Young LH. Vogt-Koyanagi-Harada disease. Semin Ophthalmol. 2005 Jul-Sep;20(3):183-90.25. Keino H, Goto H, Usui M. Sunset glow fundus in Vogt-Koyanagi-Harada disease with or without chronic ocular inflammation. Graefes Arch Clin Exp Ophthalmol. 2002 Oct;240(10):878-82. 26. Bonfioli AA, Curi AL, Orefice F. Fuchs’ heterochromic cyclitis. Semin Ophthalmol. 2005 Jul-Sep;20(3):143-6.27. Romero CP, Urzúa SC, Gallardo VP, et al. Ocular syphilis: presentation of ten cases and review of the literature. Rev Chilena Infectol. 2010 Dec;27(6):525-32.28. Ho EL, Lukehart SA. Syphilis: using modern approaches to understand an old disease. J Clin Invest. 2011 Dec;121(12):4584-92.29. Abu El-Asrar AM, Abouammoh M, Al-Mezaine HS. Tuberculous uveitis. Middle East Afr J Ophthalmol. 2009 Oct;16(4):188-201.30. How many people get Lyme disease? Centers for Disease Control and Prevention website. Updated: August 23, 2013. Available at: www.cdc.gov/lyme/stats/humanCases.html.31. Mikkilä HO, Seppälä IJ, Viljanen MK, et al. The expanding clinical spectrum of ocular lyme borreliosis. Ophthalmology. Mar 2000;107(3):581-7.32. Vasconcelos-Santos DV. Ocular manifestations of systemic disease: toxoplasmosis. Curr Opin Ophthalmol. 2012 Nov;23(6):543-50.33. Prasad AG, Van Gelder RN. Presumed ocular histoplasmosis syndrome. Curr Opin Ophthalmol. 2005 Dec;16(6):364-8.34. Foster CS, Davanzo R, Flynn TE, et al. Durezol (difluprednate ophthalmic emulsion 0.05%) compared with Pred Forte 1% ophthalmic suspension in the treatment of endogenous anterior uveitis. J Ocul Pharmacol Ther. 2010 Oct;26(5):475-83.35. Controlled evaluation of loteprednol etabonate and prednisolone acetate in the treatment of acute anterior uveitis. Loteprednol Etabonate US Uveitis Study Group. Am J Ophthalmol. 1999 May;127(5):537-44.36. Degenring RF, Jonas JB. Intravitreal injection of triamcinolone acetonide as treatment for chronic uveitis. Br J Ophthalmol. 2003 March; 87(3): 361.37. Kok H, Lau C, Maycock N, et al. Outcome of intravitreal triamcinolone in uveitis. Ophthalmology. 2005 Nov;112(11):1916-21.38. Samson CM, Waheed N, Baltatzis S, Foster CS. Methotrexate therapy for chronic noninfectious uveitis: analysis of a case series of 160 patients. Ophthalmology. 2001 Jun;108(6):1134-9.39. Fortuna E, Cervantes-Castañeda RA, Bhat P, et al. Flare-up rates with bimatoprost therapy in uveitic glaucoma. Am J Ophthalmol. 2008 Dec;146(6):876-82.40. Chang JH, McCluskey P, Missotten T, et al. Use of ocular hypotensive prostaglandin analogues in patients with uveitis: does their use increase anterior uveitis and cystoid macular oedema? Br J Ophthalmol. 2008 Jul;92(7):916-21.
