Dr. Kerr first called the phenomenon shrinkage necrosis because the dying cells somehow convert themselves into small round masses of cytoplasm, often containing tiny specks of condensed nuclear chromatin. (See Apoptosis vs. NecrosisWhats The Difference? Page 50.) These masses have their organelles intact and are eventually phag-ocytized and eaten by nearby cells, leaving no trace of the inflammation that accompanies classic necrotic cell death. The cells simply fold up and die, seemingly of their own accord.
Today, apoptosissometimes called programmed cell deathis one of hottest areas of study in cellular biology. Why? Although there are countless ways to kill healthy functioning cells, in apoptosis, many of the most universal pathologies act through common pathways to do so. This raises the hope that by modulating apoptosis and the common pathways that lead to it, a wide variety of diseasessuch as auto-immune disorders, cancer, neurodegenerative diseases and moremay be treated.
Here, well discuss how apoptosis is believed to occur in glaucoma. Also, well look at the treatments under investigation to inhibit this mechanism in optic neuropathies.
An Overview
Groundbreaking studies, such as the Ocular Hypertension Treatment Study (OHTS), the Early Manifest Glaucoma Trial (EMGT) and the Advanced Glaucoma Intervention Study (AGIS), have shown elevated intraocular pressure (IOP) to be the primary factor in glaucoma. Low-ering IOP, these studies say, prolongs ganglion cell survival and delays the onset and progression of the disease. However, since up to one-third of glaucoma patients have normal-tension glaucoma and some patients whose IOPs have been adequately lowered continue to show disease progression, high IOP may not be the only cause.
Whether the cause is mechanical trauma, ischemia, hypoxia or a combination of these and other unknown factors, elevated IOP may get the ball rolling. The lamina cribrosa becomes deformed, directly encroaching on ganglion cells and blood vessels. Blockage of blood vessels has been shown to reduce or stop axonal transport from the brain to ganglion cells, depriving retinal ganglion cells (RGCs) of much needed neuro-trophins. (Neurotrophins are small peptides that attach themselves to neuronal receptors and initiate cascades of enzymes that maintain the cells homeostasis.)
Brain-derived neurotrophic growth factor (BDNF) is a neuro-trophin that RGCs depend on. Without this vital nutrient (and/or others like it), the cell does not receive the signals it needs to live. Consequently, the cell begins to express its genetic potential for apoptosis. Enzymes inside the cell, called caspases, are activated and begin to cleave to proteins, destroying the cell from within. Eventually, the cell ingests its own DNA along with all its structural proteins and phagocytizes itself. (See Miscon-ceptions About Apoptosis. Page 52.)
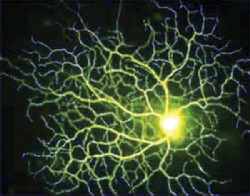 |
| Death of retinal ganglion cells (RGCs) may be a linchpin to apoptosis. |
Gene expression is at the heart of the apoptotic process. The mitochondria of healthy cells express the protein Bcl-2 on their surface. Bcl-2 and similar proteins are known to inhibit apoptosis by halting intermediate proteins that activate caspases, the main agents of internal cell destruction. Bax, is one such protein known to induce apoptosis.
A primary genetic regulator of apoptosis is the tumor suppression protein p53. Damage to DNA causes cells to increase their production of this protein. Defects in p53 are seen in cancer cellspotentially lethal cells that in a healthy organism would generate internal orders to commit suicide. This gene functions as a transcription factor that can promote the expression of the Bax protein and inhibit the expression of the Bcl-2 protein. Upregulations in the Bcl-2 protein have resulted in a 50% increase in the number of RGCs in rat models.2
Several other genes have been found to promote or inhibit apoptosis. Aside from destroying cells during the apoptotic process, caspases also proliferate important apoptotic signalers. Caspases can be switched on by increased intracellular calcium, free radicals and aden-osine 35-cyclic phosphate (cyclic
AMP).3 Compounds that show promise in increasing gene expression to prevent apoptosis (i.e., potential gene therapy agents) include deprenyl, flunarizine and aurintricarboxylic acid.3
harm healthy neurons that are located near the cells undergoing apoptosis.4
The extracellular space near these areas shows high concentrations of toxins and free radicals (molecules with an unsatisfied electron valence pair that makes them eager to bond with other molecules), depletion of growth factors and alterations in the immune systemall elements that can lead to additional cells undergoing apoptosis. Then, the process repeats itself. Thus, the area near the initial insult becomes a hostile environment in which an ongoing feedback loop of destruction con-tinues to kill cells, possibly for years after the initial insult has been alleviated.
The degree of severity seen in secondary degeneration is dependent on the severity of the initial insult. In other words, the worse the elevated IOP, the worse the secondary degeneration and subsequent feedback loop.3 Researchers theorize that the apoptotic process goes a long way in explaining two commonly observed aspects of glaucoma management:
Glaucoma patients initially diagnosed with severe optic nerve head damage and visual field loss are more likely to suffer disease progression even when brought to the same or lower levels of IOP as those who did not present with field loss upon diagnosis.
Progression continues unabated in some glaucoma patients even when their IOP has been lowered.3
|
Apoptosis vs. NecrosisWhats the Difference? |
| Contrasting apoptosis to necrosisthe conventional way cells die when exposed to mechanical damage or toxic chemicalsfacilitates the understanding of apoptosis. Apoptosis is orderly and neat; necrosis is messy and chaotic. In apoptosis cells shrink; in necrosis they swell. In apoptosis, the cells contents break down into small membrane-wrapped fragments that are engulfed by surrounding cells, causing no inflammation; in necrosis, the dying cell spills its contents into the extracellular environment, causing inflammation. Both mechanisms represent two opposite poles in a large spectrum of the way cells die. The reality often falls somewhere in between. For example, when brain cells die after a stroke, caspases, DNA fragmentation and chromosome condensation play a role, indica-ting an apoptotic process. On the other hand, the destroyed cellular contents are not wrapped in neat little membrane packages, and some of the enzymes involved do not play a role in apoptosis, indicating a more necrotic process. Ophthalmic researchers speculate that these hybrid forms of cell death may occur when the elevated intraocular pressure is so severe that it hinders the apoptotic process. Or, these hybrid forms may occur simply because vastly different cells (e.g., neurons, lymphocytes, hepatocytes, etc.) die in vastly different ways.5 |
Neuroprotection, the act of targeting these dying cells as a therapy, may be effective even if the causal factor is unknown, says James C. Tsai, M.D., chief of the division of glaucoma at Columbia University. In other words, what caused the diseasewhether ocular hypertension, ischemia, genetic factors or some unknown agentwould no longer matter if the end-game process by which the RGCs perish could be halted or retarded.
N-methyl-D-aspartate (NMDA) antagonists. These prevent overstimulation of the NMDA receptor, thus protecting the cell from an excessive influx of calcium. Too much calcium, which can be caused by excessive levels of extracellular glutamate (exotoxicity), leads to the production of free radicals, an early trigger of the apoptotic process. Like apoptosis, free radicals play a role in healthy cell function but have been implicated in pathology as well. One theory holds free radicals primarily responsible for the deleterious effects of aging. Normal free radical production is particularly high in the retina because the retina has an extremely high metabolic rate. In optic neuritis, immune cells produce toxic levels of glutamate, free radicals and other noxious agents.3
Memantine falls into the NMDA antagonist category of drug. When introduced systemically to retinal ischemia in rat models, memantine reduced RGC loss.3 In October 2003, the FDA approved Namenda (memantine, Forest Laboratories) for Alzheimers disease.
But, there is little enthusiasm for memantine as a glaucoma medication. The problem is, this is a systemic drug, says Ian B. Gaddie, O.D., in private practice in Louis-ville, Ky. It targets other cells as well as the ones in the eye.
So, until a therapy that targets only the eye is introduced, promoting memantine as an off-label glaucoma therapy may prove difficult. Still, Dr. Gaddie adds that he has had some anecdotal success using memantine with other more acute optic neuropathies.
Calcium channel blockers. These agents are used to treat cardiovascular disease. But, calcium channel blockers may also have a therapeutic effect on normal tension glaucoma patients, especially in those with blood flow problems. Retrospective studies have found that glaucoma patients who happened to be taking calcium channel blockers showed a decrease of disease progression compared with those who were not taking the cardiovascular drugs.7 Whether the benefit provided by these drugs results from inhibition of vaso-spasm, increased optic nerve blood flow or neuroprotection is not known. However, some evidence suggests that calcium channel blockers may prevent apoptosis by limiting calcium entry into cells.3
The glaucoma drug Betoptic (betaxolol, Alcon) may have an ability to block calcium activity, as may Alphagan (brimonidine, Aller-gan). Betoptic may protect cells from elevated glutamate levels, and Alphagan seems to increase cells BDNF expression in animal models.8,3
|
Misconceptions About Apoptosis |
| A lack of nutrients, such as BDNF, is not the only trigger of apoptosis. Cells may receive information signalerssuch as ultraviolet light, X-rays, chemotherapeutic drugs and the accumulation of proteins that have failed to fold into their proper tertiary structurethat prompt the death process. Also, certain molecules that bind to the cells surface receptorssuch as tumor necrosis factor-alpha (TNF-alpha), lymphotoxin (TNF-beta), Fas ligand (FasL) and Fas (also known as CD95)can start the process. Finally, cells can commit suicide based on internal signals, which occur, for example, if the cell suffers damage from free radicals. Apoptosis is not always detrimental. For instance, programmed cell death is integral to the immune response. After lymphocytes remove a foreign body, they undergo apoptosis, leaving only a small population of memory cells in their wake. When this healthy form of apoptosis fails to occur, as it does in humans who have a rare genetic defect, a condition called autoimmune lymphoproliferative syndrome (ALPS) ensues. |
Antioxidants. Glutamate cascades produce reactive oxygen
species, a known trigger of the apoptotic process. Anti-oxidants, such as catalase, superoxide dismutase and vitamins C and E can thwart toxic agents generated during secondary degeneration.3
Nitric oxide synthase (NOS-2) inhibitors. These may play a significant role in the DNA fragmentation that occurs in apoptosis. Inhibitors of NOS-2 have been shown to protect against optic nerve head cupping in glaucoma rat models.9
Neurotrophins. These are a family of growth factors that promote neuronal health. Studies have shown that the neurotrophins BDNF, ciliary neurotrophin factor and basic fibroblastic growth factor enhance the survival rate of RGCs. Evidence shows that changes in neurotrophins and their receptors may occur in retinas exposed to high IOP, thus preventing neurotrophic rescue of ganglion cells undergoing apoptosis.10
Because glaucoma occurs slowly over a period of many years, Dr. Tsai says, neuroprotection therapy would likely be long-term, and the potential side effects of tampering with the bodys natural apoptotic processes incite considerable worry. Just as some immunosuppressive drugs leave autoimmune patients open to opportunistic patho- gens, such as tuberculosis, the consequences of long-term antiapoptotic therapy might be equally serious.
Despite the explosion of knowledge in the 40 years since Dr. Kerr made his discovery, researchers have only scratched the surface of apoptosis. In the end, apoptosis will probably turn out to be a more complicated process than any of us can imagine, Dr. Tsai says. The effort of unraveling this process, however, may be well worth it.
1. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972 Aug.;26(4):239-57.
2. Martinou JC, Dubois-Dauphin M, Staple JK, et al. Overexpression of Bcl-2 in transgenic mice protection neurons from naturally occurring cell death and experimental ischemia. Neuron 1994;13:1017-30.
3. Kaushik S, Pandav SS, Ram J. Neuroprotection in glaucoma. J Postgrad Med 2003 Jan-Mar;49(1):90-5.
4. Wein FB, Levin LA. Current understanding of neuroprotection in glaucoma. Curr Opin Ophthalmol 2002
Apr;13(2):61-7.
5. Farkas RH, Grosskreutz CL. Apoptosis, neuroprotection, and retinal ganglion cell death: an overview. Int Ophthalmol Clin 2001 Winter;41(1):111-30.
6. Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science 1995;268(5210):573-5.
7. Netland PA, Chaturvedi N, Dreyer EB. Calcium channel blockers in the management of low tension and open angle glaucoma. Am J Ophthalmol 1993 May 15;115(5):608-13.
8. Gross RL, Hensley SH, Gao F, Wu SM. Retinal ganglion cell dysfunction induced by hypoxia and glutamate: potential neuroprotective effects of beta-blockers. Surv Ophthalmol 1999 Jun;43 Suppl 1:S162-70.
9. Neufeld AH, Sawada A, Becker B. Inhibition of nitric oxide synthase-2 by aminoguanidine provides neuroprotection of retinal ganglion cells in rat model of chronic glaucoma. Proc Nat Acad Sci USA 1999;96:9944-8.
10. Rudzinski M, Wong TP, Saragovi HU. Changes in retinal expression of neurotrophins and neurotrophin receptors induced by ocular hypertension. J Neurobiol 2004 Feb 15;58(3):341-54.
