Editor's note: Readers may also be interested in a more recent article on the topic from August 2023.
Visual aura represent a type of neurologic deficit familiar to any eye care practitioner. Although classically preceding migraine or seizure, an aura, simply defined, is a symptom, not a medical condition unto itself. Likewise, terms such as visual scotomas, amaurosis fugax or transient visual obscurations also represent a disturbance of vision; however, they do not classically precede migrainous headache or cortical seizure activity and are associated with other types of pathology. Nonetheless, each term, when used in the right circumstance, may define remarkably similar visual deficits in one or both eyes.
A substantial list of differentials must be considered when a patient describes such visual disturbances, some associated with significant morbidity. Because of this, the complaint of visual aura or scotoma requires a comprehensive evaluation and should not simply be assumed to be migrainous (a diagnosis of exclusion). An understanding of the different types of aura and scotomas and how they present allows eye care practitioners to differentiate causes and order testing appropriately for potentially very different pathologies.
 | |
| With scintillating, or fortification, scotomas, the central scotoma is bordered by a crescent of shimmering zigzags. |
Presentation and Pathogenesis
Visual auras or scotomas are not blur. A visual aura is a transient or longstanding visual perceptual disturbance experienced with migraine or seizure that may originate from the retina or the occipital cortex. Visual changes described by patients are often referred to as blur, a word abused by patients as frequently as the word “dizzy.” Blur has different connotative meanings to patients. Aura can be defined as either positive (seeing something that is not there) or negative (not seeing something that is there). Furthermore, an actual image may be adulterated (appears larger, perseverates, etc.). Visual auras may be transient (e.g., a few seconds) or longstanding (perhaps for months) and, importantly, they may be accompanied by headaches or other types of aura such as vertigo, numbness, tingling or aphasia.
The definition of visual scotoma is similar to that of visual aura. The differentials for scotoma likewise include migraine and seizure, but the term is more appropriately linked to ischemia, retinal degenerations and inflammations, paraneoplastic syndromes and other neurologic disorders. For visual scotomas, the primary pathogenesis may occur at the level of the receptors, retinal arterial tree, short posterior ciliary arteries, ophthalmic artery, optic nerve, carotid artery, vertebrobasilar artery or cerebral hemisphere.
Migrainous types of aura actually involve no detectable tissue pathology as well as little or no expectation of permanent deficit; however, migraine with aura has demonstrated increased risk of stroke.1 Ischemic causes, provided the ischemic threshold is not significantly surpassed, enjoy total recovery. Other causes of visual scotoma may be self-limited or require significant intervention to prevent further morbidity or mortality.
Retinal Causes
Aura that originates in the retina will present solely as an unformed scotoma or visual defect that is either positive or negative. At the retinal level, formed images are not possible.2 Although technically aura may include macropsia or micropsia, at the level of the retina this would occur with specific and typically demonstrable changes such as macular edema or cellophane maculopathy, and it would not be transient in nature.3 Transient causes of micropsia or macropsia otherwise occur cortically.
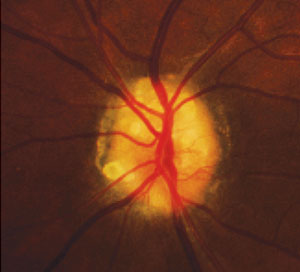 | |
| Optic nerve drusen visible on the surface of the nerve in a 32-year-old female. Photo: Denise Goodwin, OD. |
Aura at the retinal level is likely to be unilateral in presentation, but this can be difficult to elucidate from the patient’s history. Unilaterality is practically essential to attribute aura to the retina. On average, retinal aura lasts up to one hour and is most commonly embolic and rarely migrainous.
A negative visual aura deemed secondary to embolization is commonly referred to as amaurosis fugax. The Amaurosis Fugax Study Group has defined five distinct classes of transient monocular blindness based on their supposed cause: embolic, hemodynamic, ocular, neurologic and idiopathic.4 The absence of vision may or may not progress across the visual field. Retinal migraine may result in the same type of visual deficit (negative aura); however, positive scotoma or blindness is also possible. Note that retinal migraines are often, but not always, associated with headache on the same side as the visual deficit within an hour. Embolic events also may or may not be associated with headache.
Retinal ischemic events are more likely in older persons with a history of cardiovascular disease/hypertension.5 To some extent, coagulopathies or hyperviscosity syndromes may also be complicit and require consideration if a source of emboli is not identified.6 Younger patients with no history of cardiovascular disease are more likely suffering from migraine. Because monocular transient vision loss localized to the retina (classically referred to as amaurosis fugax) may have many causes, meticulous case history is important. It is critical that patients and their internist be counseled by the eye care provider regarding appropriate testing to assist in diagnosis from a large potential differential. Again, note that migraine is a diagnosis of exclusion.
Non-migrainous scotomas associated with retina may also occur at the level of the photoreceptors/RPE. The retina in a state of rest is depolarized. The state of depolarization requires energy. RPE disease in particular may interfere with energy production, which may result in localized areas of constant hyperpolarization of the affected rods and cones. As a result, a constant twinkling or sparkling scotoma may occur and can last for months or years.7 Positive continuous sparkling scotomas have been reported with conditions such as cancer-associated retinopathy, retinitis pigmentosa, or other retinal degenerations/inflammations such as multiple evanescent white dot syndrome or idiopathic blind spot enlargement.8-10
Often, retinal disorders will reveal themselves ophthalmoscopically; however, definitive dysfunction at the level of the RPE/photoreceptors may require multifocal ERG testing because fundus appearance as well as fluorescein angiography may be normal. Cancer-associated retinopathy (CAR) may result in the perception of swirling clouds of smoke and occasional dim flashes of light.11 Ophthalmoscopically, the fundus appearance early on is normal, but ERG testing may reveal significant dysfunction of rods and cones.
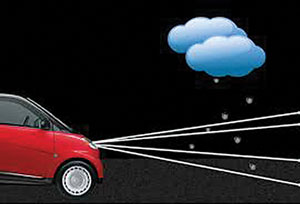 | |
| Positive scotomas are often described as snow falling through a beam of light. |
CAR is a paraneoplastic syndrome in which self-antibodies are directed toward the neoplasm but also attack specific sites in the retina, eventually resulting in arteriole attenuation, RPE mottling and disc pallor. It is most commonly associated with small-cell lung carcinoma, but has been described with other malignancies such as breast, gynecologic and prostate cancers. Chest or full-body CT scanning may be suggested in otherwise healthy patients to reveal tumor formation.
Patients with such disorders affecting cone function tend to see worse in bright light (hemeralopia), which often is clinically counterintuitive. Patient complaints of glare and (acquired) photophobia are common with cone disease. Rod disease, however, is associated with night vision difficulties (nyctalopia). Patients complaining about difficulties with both day and nighttime vision require multifocal ERG testing, which may reveal dysfunction even with the absence of visual aura. Photopsias are simply related to vitreoretinal interaction.
Optic Nerve Disease
At the level of the optic nerve, non-migrainous vision loss may occur with disorders associated with central retinal artery or short posterior artery disease. In addition, optic nerve drusen or papilledema may also be associated with aura.
As a rule, optic nerve disease produces unformed scotomas that are negative and rarely positive.2 Embolic disease blocking the central retinal artery or immediately at the bifurcation produces a negative scotoma that is either diffuse or altitudinal, respectively. The deficit may occur for seconds or up to 20 to 30 minutes. Rarely, the aura is positive and unlikely to march. Although tempting to categorize the attacks of amaurosis fugax as embolic, eye care providers must always keep in mind arteritis, where the pathogenesis may involve the ophthalmic artery or any of its branches.
Cardiac, carotid and other studies should be accompanied by a sed rate and C-reactive protein, particularly in patients over 50.12 The intention is to rule out arteritis and the potential for arteritic ischemic optic neuropathy. Short posterior ciliary disease is typically inflammatory because these vessels are not anatomically susceptible to embolic disease.13 Non-arteritic disease resulting in anterior ischemic optic neuropathy is believed to be secondary to arteriolarsclerotic disease or even possibly vascular dysregulation; however, the exact pathophysiology is unknown.14,15
Patients strongly suspected of temporal arteritis should immediately be placed on 60mg to 80mg of prednisone orally and sent to a lab for sed rate and C-reactive protein testing. Laboratory testing will not be affected for several days after initiation of prednisone therapy and will not reveal a false negative sed rate or C-reactive protein.16 Prednisone therapy is provided with the intention of preventing arteritic ischemic optic neuropathy and immediate blindness, which is a real consequence of arteritis and may occur at any time. Arteritis is one of the more common causes of preventable vision loss if detected and should always be suspected in patients with transient central, altitudinal vision loss or both.
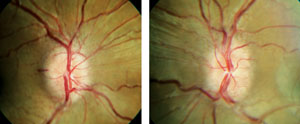 | |
| Note the bilateral, symmetric appearance of papilledema. Photo: Denise Goodwin, OD. |
Congestion of the optic nerve head may also result in transient dimming of vision, particularly with postural changes. Optic nerve drusen are space-occupying lesions believed to result in some degree of disc congestion that may produce defined persistent field defects, negative scotomas or both that may vary considerably (10 to 15 seconds up to a few hours) and are often precipitated with postural changes.17
Disc drusen are often misdiagnosed as papilledema because their chairside evaluation often resembles papilledema. Clues to diagnosis of disc drusen include the presence of a spontaneous venous pulsation, anomalous branching of arterials (trifurcations), as well as peripapillary pigmentary changes. Hemorrhages may be present and are deeper and concentric. Autofluorescence of disc drusen may be demonstrated with blue filter photography. Buried drusen are best visualized with ultrasound to reveal hyaline bodies within the nerve head; non-contrast CT is also useful, as is OCT, to differentiate optic disc drusen from papilledema.18
Although possessing an entirely different pathogenesis, papilledema may also result in negative visual scotomas (seconds to hours) that may also occur with postural changes in one or both eyes, similar to disc drusen.19
Patients with genuine papilledema will classically present with bilateral swollen or elevated optic discs with indistinct disc margins, although unilateral papilledema with opening pressures as high as 350mm have been recorded, confounding the diagnosis.20 Papilledema patients may be entirely asymptomatic, much like disc drusen patients, revealing no visual or significant field changes. Both papilledema and disc drusen patients may possess enlargement of the blind spot in both eyes, so this is not a differentiating characteristic.21,22 Typically, patients with papilledema will demonstrate no spontaneous venous pulsation because of increased cerebrospinal pressure (typically > 250mm H2O). When questioned, patients may be inclined to describe a recent history of headaches, particularly upon awakening.23 Hemorrhages, if present, tend to be superficial in the nerve fiber layer, and venules may reveal passive congestion.
Papilledema is considered one of the true ocular emergencies and patients should be transported directly to the hospital if there is any suspicion. Other compartmental or “hydraulic” pathologies resulting in compression at the apex of the orbit may also result in postural visual obscurations; therefore, orbital disease should also be kept in mind.24
Vertebrobasilar Dysfunction
Transient ischemia affecting the vertebrobasilar circulation tends to produce bilateral negative scotomas, but occasionally a patient may present with a positive scotoma. The positive visual phenomenon is often described as “snow falling through the beam of a headlight.” Like other transient ischemic attacks, the visual phenomenon may last for several minutes or longer and may be associated with other vertebrobasilar signs such as reduced awareness, diplopia, tinnitus, vertigo or dysarthria.7
Although migrainous episodes may be associated with the same deficits, like most migrainous syndromes, headache will follow in less than 60 minutes and any deficits will entirely resolve within 60 minutes. The visual deficit is still likely some type of scintillating or fortification scotoma, even if migrainous. Because of the similarity of symptoms and the potential for recovery with either mechanism, neurologic and cardiovascular evaluation is warranted with initial onset.
Cerebral Hemisphere Involvement
Most typically, visual aura that occur as a result of cerebral disease are embolic, migrainous or seizure-related. Cortical aura will be bilateral and may last anywhere from seconds up to an hour. Depending on the location (parietal, temporal or occipital), the aura will occupy that portion of the visual field commensurate with the affected lobe.
Once again, ischemic events are likely to produce negative scotoma or aura and not march. Migrainous aura are more likely to be positive (often described as scintillating) and as a rule will march. These visual aura may or may not be accompanied by headache. In the case of ischemia, headache typically occurs during onset of the aura and the visual deficit is typically negative. With migraine, the aura is accompanied or followed within 60 minutes by headache.24 If visual association areas (i.e., higher vision centers) are affected, potential phenomena that may occur include pallinopsia, macropsia, micropsia and formed visual hallucinations. Seizures affecting the occipital or sometimes the temporal lobe may also cause unformed visual hallucinations (colored circles), but may also result in formed visual hallucinations.7 Seizure activity is likely to be associated with other seizure phenomena, such as eye deviation or rapid blinking.
Besides headache, other neurologic deficits may accompany visual aura. With migraine, waves of depression that spread out of the visual cortex will typically proceed anteriorly, affecting sensation or motor strength. Each new deficit in succession may last up to 60 minutes. Therefore, the initial visual aura may be followed by parietal involvement (numbness and tingling) then frontal involvement (motor weakness). This process would be expected to take up to 180 minutes (3x60). As is typical with migraine, the deficits are expected to march and headache may occur anytime within 60 minutes after the onset of the first aura. Classically, ischemic events result in no marching and the headache is most likely to occur at the onset of vision loss. One exception to conventional visual aura is the complication of migraine “persistent aura without infarction.” In this instance, persistent visual aura—typically bilateral—will remain for at least one week and possibly months or years without evidence of ischemic injury.25 Confirmation is determined with imaging studies, which remain negative.
Dr. Banyas practices in a corporate optometry setting, and provides eye care to over 15 nursing facilities in the greater Pittsburgh area.
1. deFalco FA. Migraine with aura: Which patients are most at risk of stroke? Neurol Sci. 2015 May:36 Suppl 1:57-60.2. Ettinger AB, Veisbrot DM, eds. Neurologic Differential Diagnosis: A Case-based Approach. Cambridge, NY: Cambridge University Press; 2014.
3. Wiecek E, Lashkari K, Dakin S, Bex P. Novel quantitative assessment of metamorphopsia in maculopathy. Invest Ophthalmol Vis Sci. 2015 Jan:56(1):444-504.
4. The Amaurosis Fugax Study Group. Current management of amaurosis fugax. Stroke. 1990 Feb;21(2):210-8.
5. Callizo J, Feltgen N, Pantenburg S, et al. Cardiovascular risk factors in central retinal artery occlusion: results of a prospective and standardized medical examination. Ophthalmol. July 9 [E Pub ahead of print].
6. Schockman S, Glueck CJ, Hutchins RK, et al. Diagnostic ramifications of ocular vascular occlusion as a first thrombotic event associated with factor V Leiden and prothrombin gene heterozygosity. Clin Ophthalmol. 2015 April 3;9:591-600.
7. Purvin VA, Kawasaki A. Common Neuro-Ophthalmic Pitfalls. Cambridge, NY: Cambridge University Press; 2009.
8. Crawford C, Igboeli O. A review of inflammatory chorioretinopathies: The white dot syndromes. ISRN Inflammation. 783190. [E Pub].
9. Gass JD. Overlap among acute idiopathic blind spot enlargement syndrome and other conditions. Arch Ophthalmol. 2001;119:1729-30.
10. Jampol LM, Sieving PA, Pugh D, et al. Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol. 1984;102:671-4.
11. Jacobsen DM, Pomeranz HD. Paraneoplastic diseases of neuro-ophthalmic interest. In: Nj Newman, V Biousse, JB Kerrison, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 6th ed. Volume 2. Philadelphia: Lippincott; 2005.
12. Parikh M, Miller NR, Lee AG, et al. Prevalence of a normal C-reactive protein with an elevated erythrocyte sedimentation rate in biopsy-proven giant cell arteritis. Ophthalmol. 2006 Oct;113(10):1842-5.
13. Barash P, Cullen B, Stoelting R, et al. Clinical Anesthesia, 6th Edition, Section VII. Philadelphia: Lippincott, Williams and Wilkins; 2009.
14. Arnold AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol. 2003;23(2):157-163.
15. Collignon-Robe NJ, Feke GT, Rizzo JF. Optic nerve head circulation in nonarteritic anterior ischemic optic neuropathy and optic neuritis. Ophthalmology. 2004;111(9):1663-72.
16. Fraunfelder FT, Roy FH. Current Ocular Therapy 2. Philadelphia: W.B Saunders; 1985.
17. Knight CL, Hoyt WF. Monocular blindness from drusen of the optic disc. Am J Ophthalmol. 1972;73:890-4.
18. Sarac O, Tasci YY, Gurdal C, Can I. Differentiation of optic disc edema from optic nerve head drusen with spectral domain optical coherence tomography. J Neuroophthalmol. 2012 Sep;32(3):207-11.
19. Miller N, Subramanian P, Patel V. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, the essentials, 3rd Ed. Wolters Kluwer; 2016.
20. Brosh K, Strassman I. Unilateral papilledema in pseudotumor cerebri. Semin Ophthalmol. 2013 July;28(4): 242-3.
21. Ford CS, Biller J, Weaver RG. Drusen-associated field defects and hemorrhages. South Med J. 1983;6(8):1060-2.
22. van Endt JJ, Wessels HA. Enlargement of the blind spot caused by papilledema. Am J Ophthalmol. 1988 Sept;106(3):373.
23. Larner, AJ. Not all morning headaches are due to brain tumors. Pract Neurol. 2009 April;9(2):80-4.
24. Rose GE. Postural visual obscurations in patients with inactive thyroid eye disease; a variant of “hydraulic” disease. Eye (Lond). 2006 Oct;20(10):1178-85.
25. Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd Edition. Cephalalgia. 2013;33(9):629-808.
