Conjunctival tumors include a spectrum of benign and malignant neoplasms.1-5 The types differ based on age and race, systemic immune status and long-term exposures. A large study of 5,002 cases from an ocular oncology center revealed that 52% were benign, 18% were premalignant and 30% were malignant (Table 1).1,2 Even though this report was from an ocular oncology center and malignancies might be over-represented, it is important for clinicians to understand the variety of conjunctival tumors.
The five most common tumors were nevus (23%), ocular surface squamous neoplasia (OSSN, 14%), primary acquired melanosis (PAM, 12%), melanoma (12%) and lymphoid tumor (9%).5 Malignant tumors were seen most often in adults and included melanoma (12%), squamous cell carcinoma (SCC, 9%), lymphoma (7%), Kaposi’s sarcoma (<1%), metastasis (<1%) and others.1 Conjunctival tumors in children demonstrate malignancy only 3% of the time.5
This review of the most common conjunctival tumors will prepare you to manage them appropriately, whether in your office or through referral.
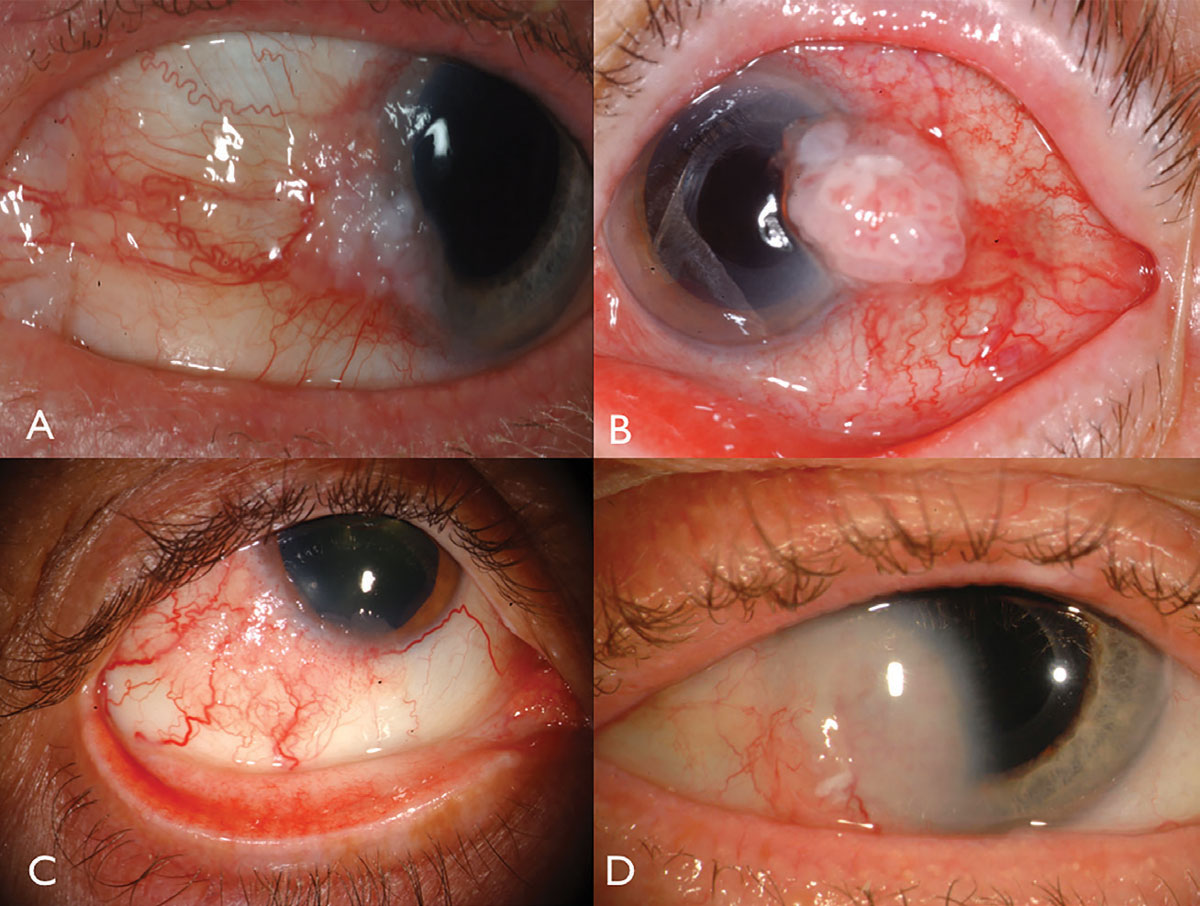 |
| Fig. 1. Limbal OSSN: with leukoplakia and corneal involvement (A), with prominent intrinsic vascularity and feeder vessels (B), in a HIV patient (C), and with deep corneal invasion requiring resection and plaque radiotherapy (D). Click image to enlarge. |
Ocular Surface Squamous Neoplasia
The general clinical term of OSSN includes a spectrum of malignancies that ranges from mild epithelial dysplastic changes, such as conjunctival intraepithelial neoplasia (CIN), to more severe invasive carcinoma that invades through the basement membrane into the substantia propria, such as squamous cell carcinoma.
Clinical features. Conjunctival OSSN classically occurs in older Caucasian males, particularly those with chronic sun exposure. In the United States, conjunctival SCC is five times more common in males and Caucasians. However, in Africa, conjunctival SCC is nearly equally common in men and women, and it occurs at a younger age than in the United States.6
Ocular surface squamous neoplasia usually presents as a unilateral, vascularized gelatinous mass, located in the sun-exposed conjunctiva at the nasal or temporal limbus (Figure 1). Overlying leukoplakia, dilated feeder vessels and foamy infiltration of the adjacent corneal epithelium can occur and can rarely invade into the globe or orbit.
 |
| Click table to enlarge. |
Predisposing factors. The most important environmental factors for OSSN include chronic sun exposure and cigarette smoke exposure. Two key host predisposing factors include fair complexion and underlying human immunodeficiency virus (HIV) and human papillomavirus.6 Patients with immune deficiency, particularly those with HIV, are at risk for OSSN and can have advanced, bilateral and invasive tumors.1 This is especially seen in Africa, where HIV is prevalent and OSSN occurs in both males and females and at a younger age.6 Other immune dysregulation can predispose a patient to OSSN, including organ transplant immunosuppression, eczema/atopy, ocular cicatricial pemphigoid, xeroderma pigmentosum and autoimmune conditions.7
Classification. The American Joint Committee on Cancer (AJCC)’s 8th edition manual provides the most recent classification for conjunctival carcinoma, including SCC and CIN (Table 2).8
Management. This involves surgical resection using the “no-touch” technique or nonsurgical therapies such as topical chemotherapy with mitomycin C (MMC) or 5-fluorouracil (5-FU), topical or injected immunotherapy with interferon alpha-2b (IFN), topical antiviral medication (cidofovir) or photodynamic therapy.7,9-11
The surgical no-touch technique involves detailed evaluation of the tumor using slit-lamp biomicroscopy to visualize all tumor margins, including bulbar, forniceal and tarsal components, to understand the entire extent of the tumor and enable the clinician to hand-draw a template recording.9 This template is then taken into the surgery to ensure the entire tumor is removed.
At the time of surgery, only the surrounding normal tissue is held with forceps, and the tumor is never touched to avoid seeding of tumor. In addition, balanced salt solution is not employed during surgery to avoid liquid-dispersion of cancer cells. Following tumor removal, closure with clean instruments is crucial. Using this technique for OSSN, tumor persistence or recurrence is found in fewer than 5% of cases.
 |
| Click table to enlarge. |
Topical chemotherapy with 5-FU or MMC is efficient in resolving the OSSN, often within two to four weeks of therapy, although a risk for stem cell deficiency exists. Our topical therapy preference is immunotherapy with IFN, as it is well tolerated with good tumor control, often over three months and with little complication and only minor follicular conjunctivitis.10,11 These medications can be locally toxic to the corneal epithelium, but less so with interferon, and patients should be followed closely while on them. If cost is a factor to the patient, topical 5-FU is the least expensive, followed by MMC and then IFN.
Conjunctival Lymphoid Tumors
Lymphoid neoplasms range from low- to high-grade tumors and arise from monoclonal proliferation of lymphocytes. The lymphoid tumors that occur in the periocular region often involve several tissues such as the conjunctiva, orbit and eyelid and are termed “ocular adnexal” lymphoid tumors, including benign reactive lymphoid hyperplasia (BRLH) and lymphoma.
BRLH and lymphoma are on opposite ends of the spectrum, with BRLH appearing clinically as a localized “salmon patch” and histopathologically benign whereas lymphoma also appears as a “salmon patch” but with more aggressive histopathologic features, with mitotic activity and classified as malignant.
Ocular adnexal lymphoid tumors are typically of B-cell origin. A multicenter study of 268 patients with conjunctival lymphoma found the four most common types included extranodal marginal zone lymphoma (ENMZL, previously termed mucosal-associated lymphoid tissue) in 68%, follicular lymphoma (FL) in 16%, mantle cell lymphoma (MCL) in 7% and diffuse large B-cell lymphoma (DLBCL) in 5%.12 Other types of conjunctival lymphoma include lymphoplasmacytic lymphoma and plasmacytoma.
Clinical features. Conjunctival lymphoma usually presents in older patients between the ages of 60 and 70. This tumor can manifest as primary lymphoma, limited to the periocular region, or as secondary lymphoma with disease elsewhere. Most primary lymphoma occurs with ENMZL and FL and secondary lymphoma with DLBCL and MCL. One analysis of 117 patients with conjunctival lymphoma found systemic involvement in 31%, most often in those with bilateral multifocal ocular adnexal lymphoma.13
Conjunctival lymphoma classically manifests as a pink salmon-colored, smooth-surfaced subconjunctival mass, sometimes with feeder vessels (Figure 2). This smooth, multilobulated mass can resemble follicular or papillary conjunctivitis. This tumor is most often located in the conjunctival fornix (44%) or midbulbar (42%) region and, rarely, in the caruncle (7%) or limbus (7%).13 In addition to the conjunctiva, lymphoma can be found infiltrating the orbit, eyelid or uvea.13 Most patients with conjunctival lymphoma do not exhibit an intraocular component, but if present, it generally resides in the uvea and not the retina or vitreous.
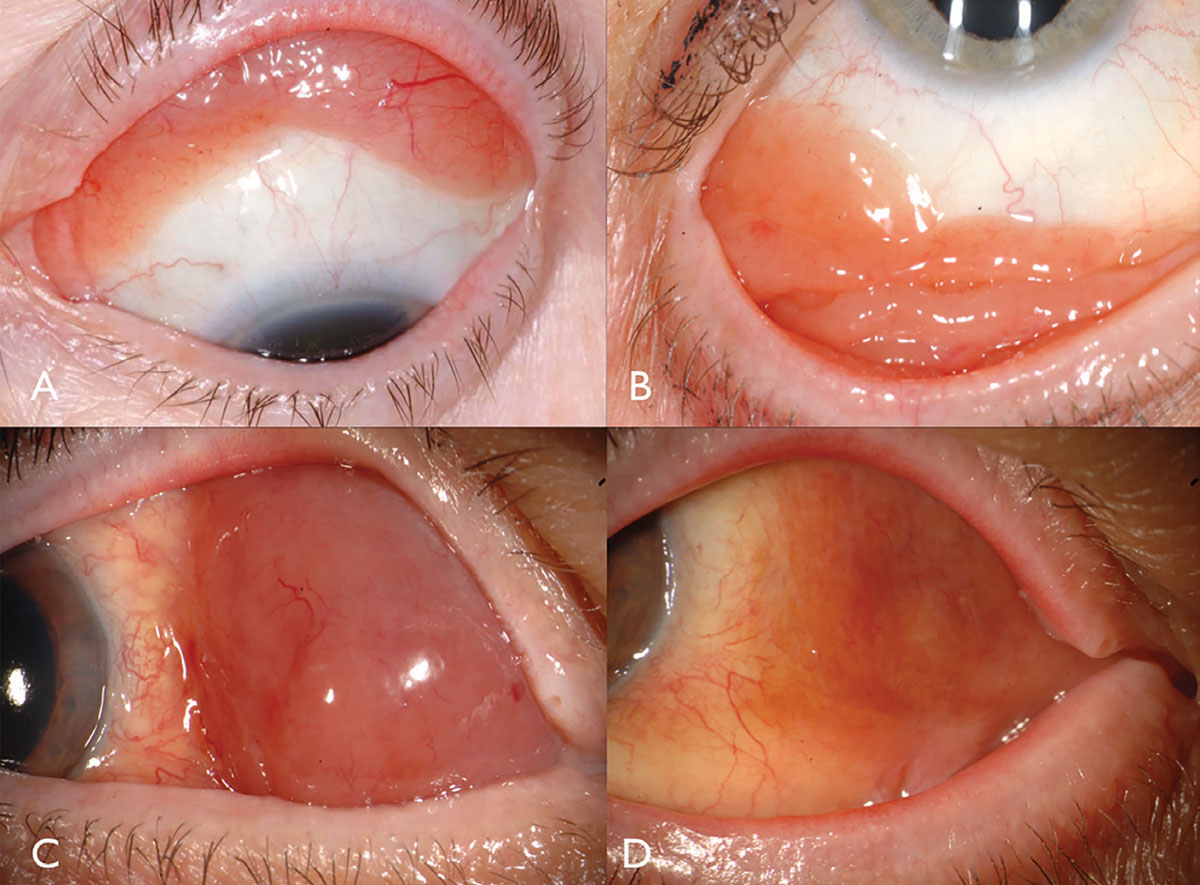 |
| Fig. 2. Conjunctival lymphoma can be salmon-pink (A) or multilobulated forniceal (B). Medial forniceal conjunctival lymphoma before (C) and after (D) ritiximab. Click image to enlarge. |
Predisposing factors. Immune dysfunction and autoimmune conditions, as well as infective etiologies such as Helicobacter pylori and Chlamydia psittaci are all predisposing factors for conjunctival lymphoma. BRLH may be a potential precursor to lymphoma and, while predominately found in adults, can occasionally occur in children.5 In fact, the younger the patient at the time of diagnosis of a conjunctival lymphoid tumor, the more likely it is BRLH and not lymphoma.
Classification. Several classifications for conjunctival lymphoma exist, including the Ann Arbor, World Health Organization and AJCC 8th edition staging (Table 3).8 The AJCC clinical staging is based on tumor location, regional lymph node and distant involvement.8
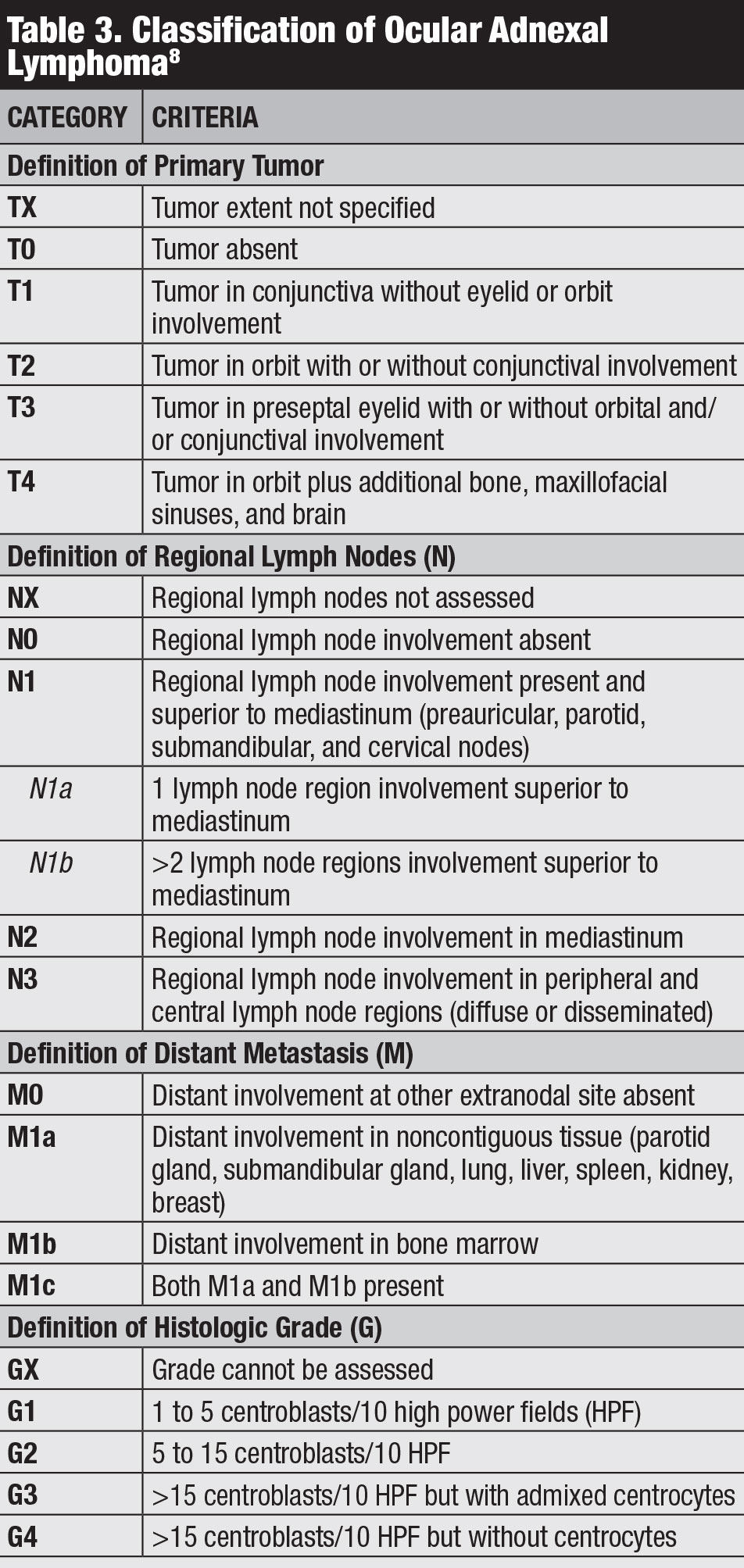 |
| Click table to enlarge. |
Management. Caring for patients with conjunctival lymphoma primarily depends on the extent of periocular involvement, systemic involvement and their general health. In patients with only conjunctival lymphoma and no systemic involvement, treatment is focused on complete surgical resection. Treatment with external beam radiotherapy or rituximab are options if the tumor is nonresectable. For those with periocular and systemic lymphoma, treatment with systemic rituximab or the addition of chemotherapy are considerations.
Systemic prognosis with conjunctival lymphoma is directly related to each subtype, as one study found the five-year survival was 97% for ENMZL, 82% for FL, 55% for DLBCL and only 9% for MCL.12
Conjunctival Melanoma
Conjunctival melanocytic tumors are unquestionably common, representing more than 50% of cases in a large series of conjunctival tumors from an ocular oncology unit.1,2 This class of melanocytic tumors includes many types such as nevus, complexion-related melanosis, PAM, secondary acquired melanosis, melanoma and metastases.1-5 On some continents where patients have dark complexions, even OSSN can appear melanocytic. Of these lesions, conjunctival nevus represents 45% and primary conjunctival melanoma represents 23% of all melanocytic tumors in an ocular oncology practice.2
In the United States, the age-adjusted incidence of conjunctival melanoma doubled between 1973 and 1999 from 0.27 per million to 0.54 per million.14,15 The incidence increased 295% in white men in the United States over the same 27-year period, especially among men aged 60 years or older.14 Researchers speculate the increasing rate may be related to ultraviolet light exposure.
Clinical features. Conjunctival melanoma is a pigmented or non-pigmented malignancy that can arise from PAM, nevus or de novo.16 Melanoma can be found on the limbal, bulbar, forniceal or palpebral conjunctiva and often demonstrates dilated, tortuous feeder and intrinsic vessels typically surrounded by flat PAM (Figure 3). In general, tumors measuring greater than 2mm in thickness are at significant risk for lymph node metastasis. Tumor invasion into the orbit is particularly serious with substantial metastatic risk.
Local tumor recurrence or new tumor is found in 50% of cases, often related to new PAM transformation. Distant metastasis—often to the preauricular, submandibular or cervical lymph node chain—is found in 25% of patients. Sentinel lymph node biopsy can help clinicians evaluate for subclinical lymph node infiltration. Multiple recurrences, especially those involving the orbit, necessitate orbital exenteration.
Predisposing factors. The most important predisposing factor for conjunctival melanoma is the presence of long-standing conjunctival nevus or PAM.16-18 When studying conjunctival melanoma origin by histopathology, researchers found the origin was PAM in 74%, de novo in 19% and nevus in 7%.16 Clinical studies estimate that one in 300 nevi develop into melanoma.17,18
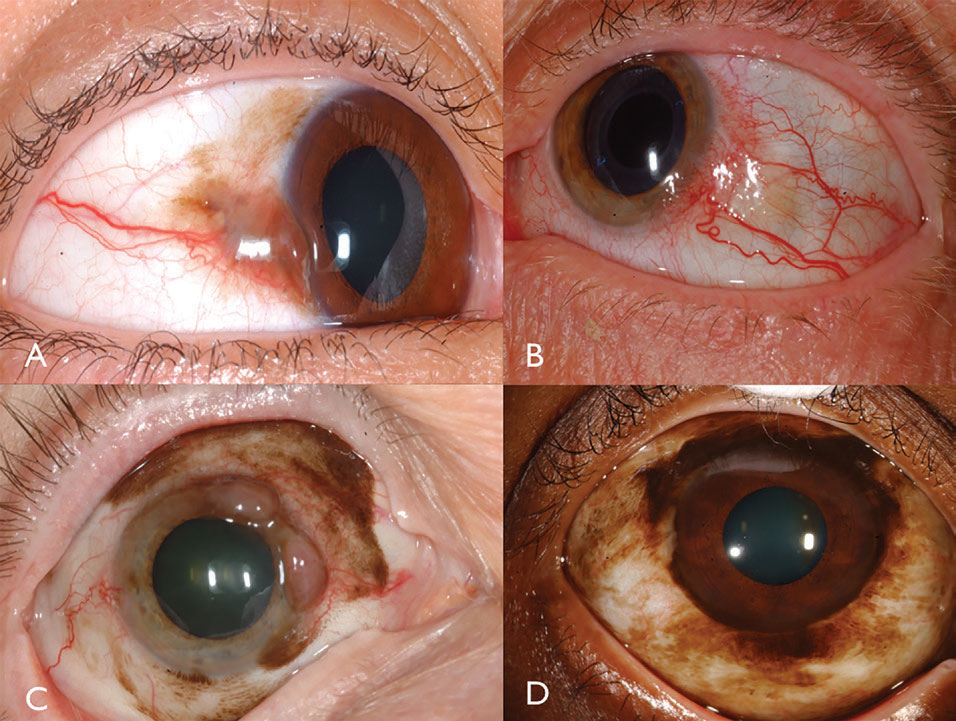 |
| Fig. 3. Pigmented conjunctival melanoma can arise from PAM (A). Non-pigmented conjunctival melanoma may have intense vascularity (B). PAM could also cause mixed pigmented/non-pigmented conjunctival melanoma (C). PAM caused limbal melanoma in this African-American patient (D). Click image to enlarge. |
A large clinical study found that the 10-year risk for PAM transformation into melanoma was about 9%, and the greater extent of PAM promoted a greater risk for transformation into melanoma.19 Hence, it is important to identify PAM and treat this condition with surgical excision, cryotherapy and even superficial keratectomy (if there is corneal involvement) with the intention of preventing melanoma.
The differentiation of conjunctival nevus from melanoma can be challenging. In a recent analysis of 510 cases of conjunctival nevus vs. melanoma in children, melanoma was more common in older children, with a relative risk (RR) of 4.80, greater tumor thickness (RR of 1.14), greater base (RR of 4.92), tumor hemorrhage (RR of 25.30) and lacking intrinsic cysts (RR of 5.06).5 The researchers assigned these features, predictive of conjunctival melanoma in children, to a mnemonic: CATCH Melanoma, representing: Children Age older, Thickness/base greater, Cyst lacking, Hemorrhage for Melanoma.5
Differentiation of PAM from melanoma can also be challenging; however, melanoma has thickness and PAM is completely flat. In an analysis of 1,224 cases of PAM vs. melanoma in all ages, melanoma with significantly greater based on median patient age (54 vs. 61 years); male sex (35% vs. 49%); location in fornix (2% vs. 6%) and tarsus (1% vs. 4%); larger median basal diameter (6mm vs. 8mm), thickness (<1mm vs. 1mm), feeder vessels (10% vs. 48%) and intrinsic vessels (4% vs. 33%); and hemorrhage (<1% vs. 3%).2
Tissue biomarkers are important for the assessment of conjunctival melanoma and include BRAF mutation, TERT promoter mutation and PTEN mutation.1 Identifying these biomarkers is critical when planning systemic therapy for treatment or prevention of metastasis, as targeted therapies against certain biomarkers are available, such as vemurafenib for BRAF-mutated malignancy.
 |
| Click table to enlarge. |
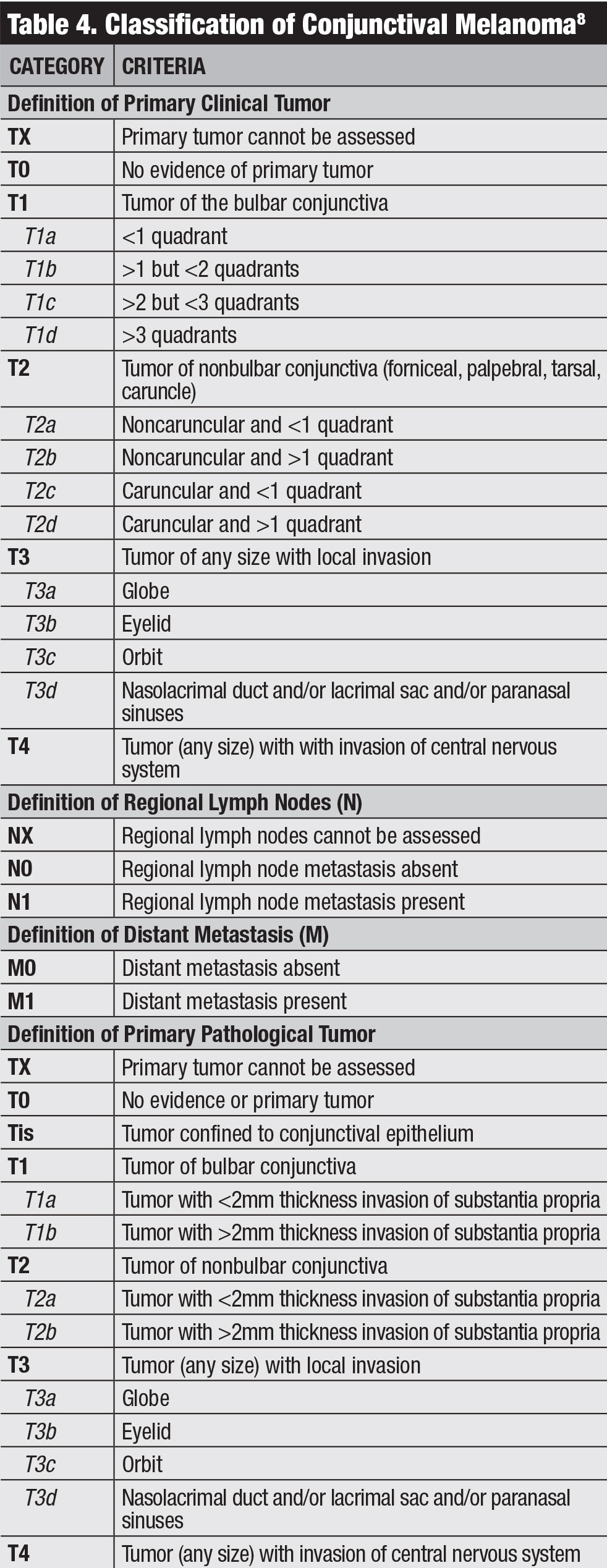
Classification. The AJCC’s clinical classification for conjunctival melanoma is based on tumor extent by quadrants, tumor location and invasive features (Table 4).8 Our team studied outcomes of conjunctival melanoma based on the AJCC 7th edition and found this staging was highly predictive of prognosis.20 Melanoma classified as T2 and T3 (compared with T1) showed significantly higher rates of local recurrence, regional lymph node metastasis, distant metastasis and death.
Management. Care for conjunctival melanoma basically involves complete surgical resection using the no-touch technique to avoid tumor seeding. The first surgery is the most important, as delicate removal of the entire tumor without tumor seeding is key to preventing future recurrences and metastases.16
Melanoma at the corneoscleral limbus is removed under the operating microscope also using the no-touch technique. The flat corneal component is removed with absolute alcohol, superficial epitheliectomy without disruption of Bowman’s membrane. The conjunctival portion is removed with 2mm to 3mm margins and released at the limbus using flat episcleral dissection. If scleral invasion is present, plaque radiotherapy is applied. All conjunctival margins are treated with double freeze-thaw cryotherapy.
Reconstruction involves primary closure techniques, rotational flap or amniotic membrane transplantation, often with symblepharon ring with amniotic membrane draping. Melanoma that extends into the orbit requires orbital exenteration or, more recently, immunotherapy with checkpoint inhibition.21
Patients with conjunctival melanoma should be monitored by an ocular oncologist for local recurrence and by a systemic melanoma oncologist for metastatic disease, particularly with regional lymph node palpation and sentinel lymph node biopsy. Metastases initially appear in the preauricular or submandibular lymph nodes, then later in the lung and brain. New evidence suggests that melanoma metastasis could be sensitive to BRAF inhibitors or immune checkpoint inhibitors.21,22
Conjunctival tumors encompass a broad spectrum of tumors. The most common malignancies include OSSN, lymphoma and melanoma. Recognizing the classic clinical features, understanding precursors and prompt and appropriate management of these malignancies are important for best patient outcomes.
Drs. Shields, Lally and Shields work in the Ocular Oncology Service at Wills Eye Hospital, Thomas Jefferson University, in Philadelphia. Support provided by Eye Tumor Research Foundation, Philadelphia.
|
1. Shields CL, Chien JL, Surakiatchanukul T, et al. Conjunctival tumors: review of clinical features, risks, biomarkers, and outcomes. The 2017 J. Donald M. Gass Lecture. Asia Pac J Ophthalmol. 2017;6:109-20. 2. Shields CL, Alset AE, Boal NS, et al. Conjunctival tumors in 5002 cases. Comparative analysis of benign versus malignant counterparts. The 2016 James D. Allen Lecture. Am J Ophthalmol. 2017;173:106-33. 3. Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol. 2004;49:3-24. 4. Grossniklaus HE, Green WR, Luckenbach M, et al. Conjunctival lesions in adults. A clinical and histopathologic review. Cornea. 1987;6:78-116. 5. Shields CL, Sioufi K, Alset AE, et al. Conjunctival tumors in children. Features differentiating benign from malignant tumors. JAMA Ophthalmol. 2017;135:215-24. 6. Gichuhi S, Sagoo MS, Weiss HA, et al. Epidemiology of ocular surface squamous neoplasia in Africa. Trop Med Int Health. 2013;18:1424-43. 7. Shields CL, Ramasubramanian A, Mellen P, Shields JA. Conjunctival squamous cell carcinoma arising in immunosuppressed patients (organ transplant, human immunodeficiency virus infection). Ophthalmology. 2011;118:2133-7. 8. Amin MB, Edge S, Greene F, et al., eds. AJCC Cancer Staging Manual. 8th ed. Chicago; Springer International Publishing: 2017. 9. Shields JA, Shields CL, De Potter PV. Surgical approach to conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol. 1997;115:808-15. 10. Shields CL, Kaliki S, Kim HJ, et al. Interferon for ocular surface squamous neoplasia in 81 cases: Outcomes based on the American Joint Committee on Cancer classification. Cornea. 2013;32(3):248-56. 11. Karp CL, Galor A, Chhabra S, et al. Subconjunctival/perilesional recombinant interferon α2b for ocular surface squamous neoplasia: a 10-year review. Ophthalmology. 2010;117(12):2241-6. 12. Kirkegaard MM, Rasmussen PK, Coupland SE, et al. Conjunctival lymphoma – An international multicenter retrospective study. JAMA Ophthalmol. 2016;134:406-14. 13. Shields CL, Shields JA, Carvalho C, et al. Conjunctival lymphoid tumors: clinical analysis of 117 cases and relationship to systemic lymphoma. Ophthalmology. 2001;108:979-84. 14. Yu GP, Hu DN, McCormick S, Finger PT. Conjunctival melanoma: Is it increasing in the United States? Am J Ophthalmol. 2003;135:800-6. 15. Tuomaala S, Kivela T. Correspondence regarding conjunctival melanoma: Is it increasing in the United States? Am J Ophthalmol. 2003;136:1189-90. 16. Shields CL, Markowitz JS, Belinsky I, et al. Conjunctival melanoma. Outcomes based on tumor origin in 382 consecutive cases. Ophthalmology. 2011;118:389-95. 17. Shields CL, Fasiuddin AF, Mashayekhi A, et al. Conjunctival nevi: clinical features and natural course in 410 consecutive patients. Arch Ophthalmol. 2004;122:167-75. 18. Gerner N, Norregaard JC, Jensen OA, Prause JU. Conjunctival naevi in Denmark 1960-1980. A 21-year follow-up study. Acta Ophthalmol Scand. 1996;74:334-7. 19. Shields JA, Shields CL, Mashayekhi A, et al. Primary acquired melanosis of the conjunctiva: risks for progression to melanoma in 311 eyes. The 2006 Lorenz E. Zimmerman lecture. Ophthalmology. 2008;115:511-9. 20. Shields CL, Kaliki S, Al-Dahmash S, et al. American Joint Committee on Cancer (AJCC) Clinical Classification predicts conjunctival melanoma outcomes. Ophthalm Plast Reconstr Surg. 2012;5:313-23. 21. Sagiv O, Thakar SD, Kandl TJ, et al. Immunotherapy with programmed cell death 1 inhibitors for 5 patients with conjunctival melanoma. JAMA Ophthalmol. 2018 Nov 1;136(11):1236-41. 22. Dalvin LA, Shields CL, Orloff M, et al. Checkpoint inhibitor immune therapy. Systemic indications and ophthalmic side effects. Retina. 2018;6:1063-78. |
