In April 2016, optometry lost a giant when the author of the seminal work Primary Care of the Posterior Segment, Larry Alexander, OD, died. In addition to being an optometric physician, author and educator at the University of Alabama Birmingham School of Optometry, Dr. Alexander was a past president of the Optometric Retina Society (ORS). That group chose to honor his legacy by accepting case reports from optometric residents across the country relating to vitreoretinal disease.
This case, selected by the ORS Awards Committee, was co-winner of the fifth annual Larry Alexander Resident Case Report Contest. The contest is sponsored by Zeiss, Heidelberg and Optos.
The purpose of this case report is to present a case of bilateral, combined hamartoma of the retina and retinal pigment epithelium (RPE) leading to a suspected diagnosis of neurofibromatosis type 2. Combined hamartomas of the retina and RPE are rare intraocular tumors characterized by the malformation of the neurosensory retina, RPE and adjacent vitreous. The majority of these lesions occur in isolation, but the association with several neurocutaneous disorders is well established. Thus, all patients with combined hamartoma of the retina and RPE require careful eye exams and detailed case history.
This case presentation discusses the clinical characteristics of combined hamartomas of the retina and RPE as well as other potential ocular manifestations of neurofibromatosis type 2 and the importance of early diagnosis of neurofibromatosis type 2.
Introduction
Combined hamartomas of the retina and retinal pigment epithelium (CHRRPE) are rare, congenital, intraocular tumors characterized by the malformation of the neurosensory retina, RPE and adjacent vitreous with disorganized glial, vascular and melanocytic tissue.1-5 On fundus examination, these lesions often appear elevated, pigmented (usually grey, white or brown, but may be orange, yellow or green) and are frequently associated with overlying glial tissue that may produce retinal distortion, wrinkling and/or striae.6-9 On OCT imaging, consistent features of CHRRPEs include varying amount of thickened retinal and pre-retinal tissue, retinal disorganization, epiretinal membranes with secondary retinal folds and striae, normal adjacent retina and no evidence of choroidal involvement.10-12 Most isolated cases of CHRRPE are unilateral. Bilateral cases of CHRRPE are suggestive of phakomatoses (neurocutaneous syndromes), notably neurofibromatosis type 2 (NF2).13
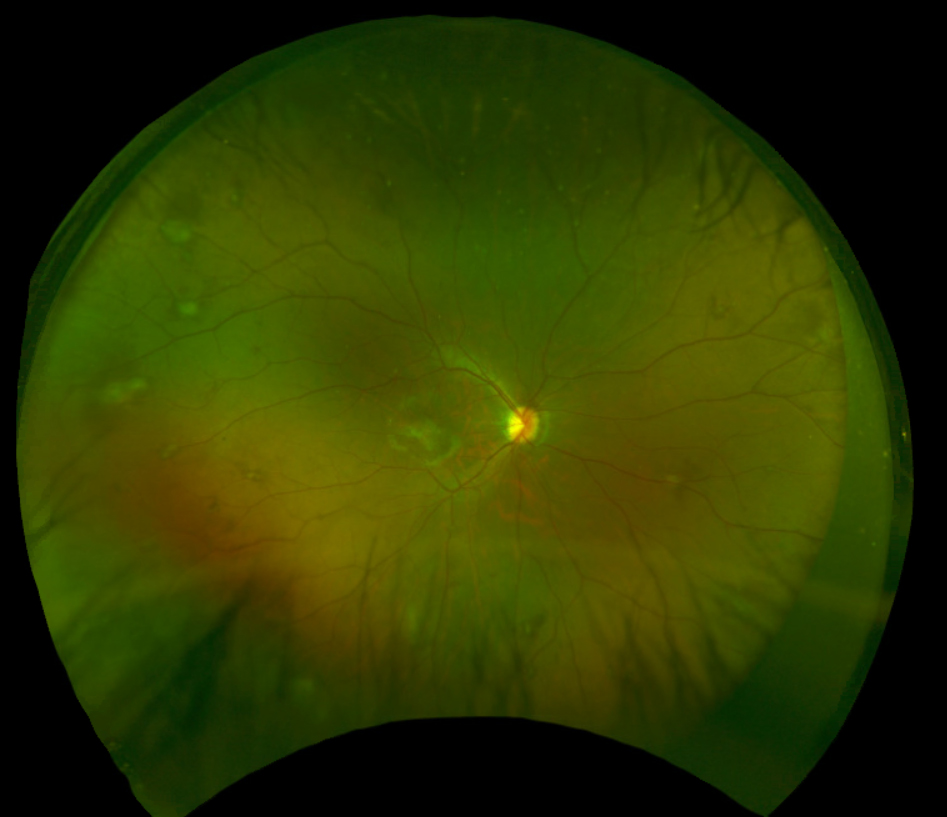 |
| Fig.1. Fundus photograph of the right eye captured with Optos. Click image to enlarge. |
Case Report
Comprehensive Eye Examination (02/25/2021)
A 12-year-old Hispanic male, accompanied by his biological mother, presented to the eye center as a new patient for a comprehensive eye examination. His chief complaint was blurry vision in the right eye more than the left eye since kindergarten. He reported stable vision in both eyes and that vision was better when he wore his glasses of six months. Squinting helped mildly, he noted. His symptoms were constant in nature. He denied any ocular pain, redness, irritation, eye crossing or eye drifting. He denied seeing any flashes of light or floaters.
The patient's last eye exam was November 2020. He reported that he was given new glasses and told to schedule an eye examination at this practice due to “tumors in his eyes” but neither he nor his mother could recall being told anything more. He denied any past ocular history of surgery or trauma. His family ocular history was negative. The patient’s medical history was unremarkable. His only active medication was a 0.3mg epinephrine auto-injector for a known allergy to bee venom, of which he reported a type 1 hypersensitivity reaction (anaphylactic shock). He had no known allergies to any systemic medications. The patient’s mother reported an uncomplicated pregnancy and birth. Social history was negative for tobacco, alcohol or recreational drug use. He was oriented to person, place and time, and his mood was appropriate.
Spectacle corrected distance visual acuity was 20/125 OD and 20/50-2 OS. The patient’s habitual single vision spectacle correction measured with lensometry was -6.50 +0.75 x 096 in the right eye and -4.75 +1.00 x 080 in the left eye. Unilateral and alternating cover testing revealed orthophoria at distance and near. The patient was only able to appreciate two out of nine circles with the stereo test. The patient was found to be suppressing his right eye with Worth 4-Dot at distance and near. Confrontation visual fields were full to finger counting in both eyes. Function of extraocular muscles were found to be smooth, accurate, full and extensive with no pain nor diplopia. Pupils were equal, round and reactive to light with no afferent pupillary defect noted.
Examination of the anterior segment using slit lamp biomicroscopy revealed normal adnxae, lashes, puncta, sclera and palpebral and bulbar conjunctiva in both eyes. The upper and lower eyelids were clean and free of debris in both eyes. All puncta were patent and normal in appearance. The lower palpebral conjunctiva was negative for both follicles and papillae with lower eyelid eversion. The corneas were intact and clear through all layers in both eyes. Both irides were flat and brown. The anterior chambers of both eyes appeared deep, with no evidence of cell or flare and the angles were estimated to be grade 4+ using the Van Herick method. An iCare tonometer measured the intraocular pressure. The intraocular pressure was 14mm Hg in the right eye and 16mm Hg in the left eye at 8:24am. The patient was dilated using one drop of 1% cyclopentolate with 2.5% phenylephrine and 1 drop of 0.5% tropicamide with 5% phenylephrine in both eyes at 9:59am.
Upon full cycloplegia and dilation, the cycloplegic refraction revealed -7.00 +1.00 x 090 OD and -5.75 +2.00 x 080 OS. The best-corrected visual acuity at distance was 20/100 OD and 20/30-2 OS. The visual acuity did not improve with a pinhole occluder over the cycloplegic refraction in either eye.
An evaluation of the posterior segment for each eye was conducted using slit lamp biomicroscopy with a 90D lens and by a binocular indirect ophthalmoscope with a 20D lens. The lens was clear in each eye. There were no white blood cells nor pigment in the anterior vitreous.
The optic nerves appeared well perfused with a slight temporal tilt and a cup-to-disc ratio of 0.25 round in both the right and left eye. The macula of the right eye was remarkable for an elevated, grey-white lesion (approximately 2DD by 2DD over the fovea) with overlying epiretinal membrane/surface wrinkling (Figure 1). The macula of the left eye appeared flat but there was an elevated, yellow-white, distinct lesion (approximately 2DD by 2DD) temporal to the fovea (approximately 2.5DD away) (Figure 2). The foveal reflex was absent in both eyes. Evaluation of the peripheral retina revealed multiple scattered, grey-white elevated lesions in both eyes.
Spectral-domain optical coherence tomography (OCT) of the retina (Heidelberg Spectralis) and fundus photography (Daytona, Optos) was ordered for both eyes. The OCT of the right eye revealed an elevated, hyperreflective mass over the fovea with prominent retinal thickening and notable disorganization of the retinal architecture, with major involvement of the inner retinal layers. An overlying epiretinal membrane with vitreomacular traction was also noted. The OCT of the left eye revealed vitreoretinal interface changes at the fovea with a distorted foveal contour (Figure 3). Additionally, the OCT of the left eye captured the edge of the previously mentioned temporal lesion, which showed similar retinal thickening/disruption. The macular thickness map of each eye revealed elevation at the fovea and over the previously mentioned lesions.
The differential diagnosis for these lesions considered at-this-time of the examination included:
- CHRRPE
- Retinal astrocytic hamartoma
- Intermediate uveitis or pars planitis
- Retinoblastoma
These retinal lesions had the classic characteristics of CHRRPEs; they were elevated, pigmented and associated with retinal disorganization with overlying glial tissue proliferation. This was observed both with posterior segment evaluation and OCT. There was an absence of choroidal involvement, retinal detachment, hemorrhage, exudation and vitreous inflammation. Thus, the patient was diagnosed with multiple bilateral CHRRPEs.
Retinal astrocytic hamartomas (RAH) are benign, glial tumors of the retinal nerve fiber layer that arise from retinal astrocytes. Classically, they appear as white, semitransparent or multinodular “mulberry appearing” tumors in the superficial retina. They may be unilateral or bilateral, solitary or multifocal, calcified or noncalcified.14-16 Characteristics features on OCT of RAH include: characteristic “moth-eaten” spaces (optically empty spaces representing intralesional calcification or intratumoral cavities), a dome shaped surface, posterior shadowing and gradual transition from normal retina into an optically hyperreflective mass with retinal disorganization and abnormal thickening of the retinal nerve fiber layer.7,16
Considering the variable presentations of RAH, this diagnosis was strongly considered. Additionally, CHRRPE and RAH share similar features with OCT studies (gradual transition from tumor tissue to adjacent normal retina, full-thickness retinal occupation and retinal disorganization). However, CHRRPE tends to have more profound pre-retinal traction and retinal disorganization.10,12,16 This patient lacked the characteristic “moth-eaten” space or dome shaped surface but did have a prominent overlying epiretinal membrane and notable retinal disorganization, consistent with a diagnosis of CHRRPE rather than RAH.
Intermediate uveitis is characterized by mild to moderate anterior segment inflammation, diffuse cell/haze, snowballs and snow-banking.17-21 The lesions of this patient were distinct and white in appearance, similar to how snowballs may present; however, snowballs are located in the vitreous and more commonly found in the inferior peripheral retina. This patient also lacked any signs of ocular inflammation; thus, this differential diagnosis was deemed unlikely.
Retinoblastoma is the most common ocular cancer in children. In 90% of cases, the diagnosis is made before five years of age, with a mean age of 18 months.17,22 The lesions of this patient did not invade the RPE or choroid and transitioned gradually into normal adjacent retina. There was also no subretinal fluid, feeder vessels, serous detachment, calcification or inflammation in either eye. Retinoblastoma was considered an unlikely diagnosis due to the patient’s older age and because the retinal lesions did not appear suspicious for malignancy. However, given that this patient was new to the practice, it was deemed critical to follow up with him to establish stability of his ocular health.
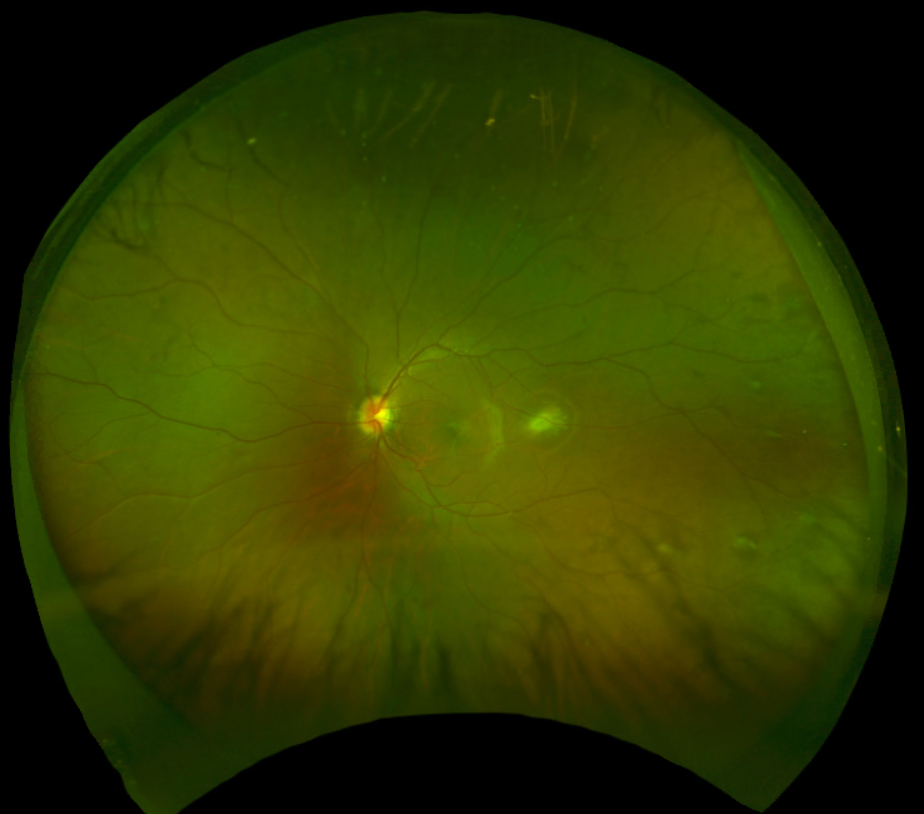 |
| Fig. 2. Fundus photograph of the left eye captured with Optos. Click image to enlarge. |
Diagnosis
This patient had no known systemic conditions per his mother. However, after further investigating his family's systemic history, it was revealed that the patient’s father was involved in an accident five years prior and subsequently suspected of having NF2 after two tumors were identified. Unfortunately, further medical workup and management was not reasonably accessible to the patient’s father, as he lacked medical insurance. The patient was identified as an NF2 suspect secondary to his remarkable ocular findings (bilateral CHRRPEs) and his positive family history. Additionally, derivational amblyopia was suspected in the right eye due to the long-standing decreased visual acuity.
A retina specialist was consulted, who agreed with the diagnosis of bilateral CHRRPE and epiretinal membranes of both eyes. The specialist was able to discuss with the patient and his mother the possible benefit of pars plana vitrectomy and membrane peel of the right eye, but the decision was ultimately made to hold off on ocular surgery, given the likely long-standing nature of his condition and the potential amblyopic component to his decreased vision. However, surgery was deemed reasonable if the patient experienced a decrease in vision with progression of the epiretinal membrane/vitreomacular traction. An updated spectacle prescription was provided to the patient.
The patient was to return to the practice for a repeated dilated fundus exam, OCT and fundus photography of both eyes. Most importantly, the patient was referred to the local neurofibromatosis clinic for further investigation and formal diagnosis. An audiology assessment and MRI of the brain with and without contrast with an internal auditory canal protocol is planned for that visit. The patient will likely be offered genetic testing at that appointment.
Discussion
Combined hamartoma of the retina and RPE are rare fundus tumors with classic clinical features. Histopathological studies of CHRRPE demonstrate marked disorganization of the architecture of the involved retina with proliferation of the RPE through the retina and along the inner retinal surface, with hyperplastic glial and vascular tissue. Historically, CHRRPE has been described as a tumor primarily arising from all retinal layers; however, the involvement of RPE has been questioned based on evidence from studies focusing on the OCT features of the lesion.23,24
The most common clinical features of CHRRPE, described by Schachat et al. in a series of 60 cases in 1984, included pigmentation (87%), elevation (80%), vascular tortuosity (93%), vitreoretinal interface changes (80%) and lipid exudates (7%).25 Uncommon associated findings include secondary macular edema, exudation, choroidal and preretinal neovascularization, retinoschisis and retinal detachment.7
It is important to acknowledge that because epiretinal membrane formation is a prominent clinical feature of CHRRPEs, overlying vitreoretinal interface disturbances with retinal traction may lead to poor vision.1,2,4 Shields et al. studied 11 patients with spectral-domain OCT; they observed epiretinal membranes with corresponding retinal folds and striae leading to retinal anatomical disorganization in all the patients.5,25 A later retrospective review by Shields et al. evaluated the visual outcome of 79 eyes with CHRRPE. They found that symptoms of reduced visual acuity and/or strabismus were present in 74% of patient.
By univariate analysis, the most important factors predictive of poor visual acuity (equal to or less than 20/200) included macular location and clock hour meridian of the tumor.4 To date, there is no highly effective method for treating CHRRPE. Vitrectomy and membrane peeling can be helpful in cases with vitreous hemorrhage and preretinal gliosis. In the rare case of neovascularization membrane formation, laser photocoagulation may be employed.26
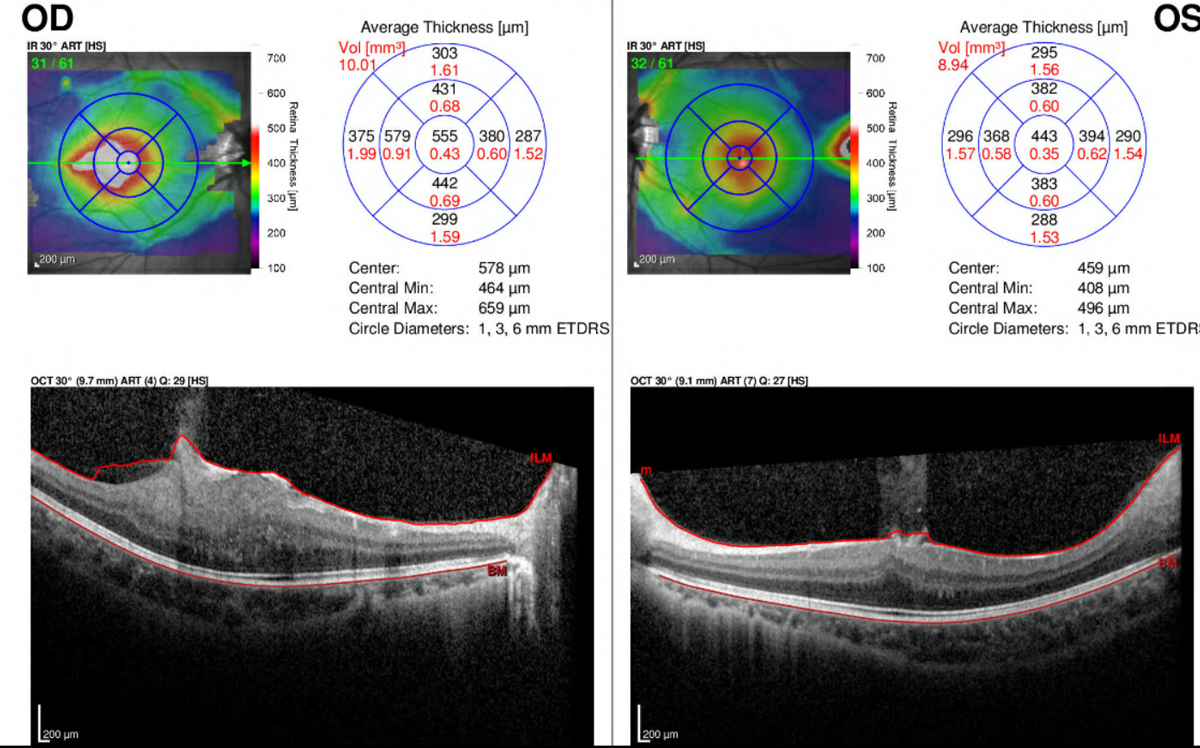 |
| Fig 3. OCT with macular thickness map of the right and left eye. Click image to enlarge. |
CHRRPE are classically recognized in young children with symptoms of strabismus or reduced visual acuity. Schachat et al. found the mean age at diagnosis was 15 years.25 This is contrasted by Shields et al. who found a mean age of diagnosis of one year (the youngest child in the series identified at two weeks of age); this is likely attributed to the tertiary nature of the practice with special interest in pediatric and adult retinal tumors.4
These lesions tend to remain relatively stable but can evolve slowly with age. Bilateral lesions are more frequently associated with phakomatoses, including NF2, neurofibromatosis type 1 (NF1), tuberous sclerosis and basal cell nevus syndrome (also referred to as Gorlin syndrome).11,27,28 Of the phakomatoses, CHRRPE are more common in patients with NF2; however, they have been reported in patients with NF1 which is a more prevalent disease, thus evaluation for both disorders has been recommended.28
Neurofibromatosis type 2 is a rare, autosomal dominant disorder characterized by formation of central nervous system tumors due to a mutation in the tumor suppressor gene NF2. This gene is located on chromosome band 22q12 and encodes a 595-amino acid protein known as merlin (a moesin-ezrin-radixin-like protein).29,30 First described in 1993, merlin is important for proper anchorage of the cytoskeleton to the cell membrane, cell membrane proteins organization and cytosolic proteins interactions.31,32 These pathways are required for proper cell growth, protein translation and cellular proliferation. Without a normal merlin protein, individuals are predisposed to tumor formation.33
Bilateral vestibular schwannomas are the hallmark of the condition (affecting 95% of individuals); however, patients often develop other nervous system tumors (schwannomas of other cranial, spinal and cutaneous nerves, meningiomas, ependymomas, astrocytomas and neurofibromas), peripheral neuropathy, cutaneous lesions and ophthalmological lesions.27,34,35 Although histologically benign, these tumors are associated with significant morbidity if multiple central nervous systems are affected.34 NF2 has wide phenotypic variability and nearly 100% penetrance by 60 years of age.36,37
The incidence of NF2 is estimated at one in 33,000 to 50,000.36 The prevalence is estimated around one in 60,000.38 Approximately 50% of cases arise from de novo mutation.39 About 10% of patients present before the age of 10 years and 18% before the age of 15 years.40 This condition is typically diagnosed in the second or third decade of life following initial symptoms of tinnitus, hearing loss and/or balance dysfunction secondary to vestibular schwannomas. Children more frequently present with visual disturbances, skin tumors, mononeuropathy (facial paresis, foot drop), symptomatic spinal cord tumors or non-vestibular intracranial tumors.35,36,41,44
Ophthalmological abnormalities are well described and present in the majority of NF2 patients. The most common ophthalmic manifestations are cataracts (60% to 81%), followed by epiretinal membranes (12% to 40%) then retinal hamartomas (CHRRPE) (6% to 22%).27,41-25 These ocular manifestations are usually present congenitally and patients may become more symptomatic over time.28,44,47 Cataracts are more commonly bilateral and of the juvenile posterior subcapsular or cortical types. Usually patients are asymptomatic, but cataract extraction is indicated in approximately 10% to 35% of these cases.30-32 Other associated ocular manifestations include strabismus (12% to 50%), amblyopia (12%), optic nerve sheath meningiomas (28%), other optic pathway tumors (10% to 27%) and extraocular movement abnormalities (10%).34,42,48 Corneal injury and/or neurotrophic keratopathy may occur following facial nerve weakness. Nystagmus has also been described as a result of peripheral vestibular dysfunction.33 It is important to consider that visual development may be compromised in these patients and careful ophthalmic examination should be performed in all patients with NF2 or suspected of NF2.
The multiple, progressive features associated with NF2 present substantial management challenges. These patients require care from a multidisciplinary team, including neurology, ophthalmology, genetics, audiology, dermatology, medical and radiation oncology, clinical nurse consultants, allied health staff and psychologists.34 A study in 2002 showed that NF2 patients who were referred for care in specialty treatment centers had a lower risk of mortality.49 Optimum management includes screening populations at risk, early diagnosis, close surveillance, comanagement with multiple health care specialists and tailoring treatment to preserve function and quality of life.50
Conclusion
Combined hamartomas of the retina and retinal pigment epithelium are rare congenital, benign intraocular tumors that are typically unilateral and remain stable over time. However, vision loss can result if there is loss of normal retinal structure/architecture due to prominent glial tissue proliferation. Once bilateral CHRRPE are confirmed, phakomatous etiologies should be considered. Neurofibromatosis type 2 is a multiple neoplasia syndrome that predisposes patients to the development of tumors in the nervous system, eyes and skin. The most common ocular manifestations include infantile cataracts, epiretinal membranes and CHRRPEs. It is important for eye care practitioners to recognize that children with NF2 will more commonly present with ocular, dermatological and/or neurological signs and require careful ophthalmic examination and comanagement. Additionally, if the patient is suspected of having NF2, prompt diagnosis and referral to a multidisciplinary clinic is warranted to reduce morbidity and mortality.
1. Gass JD. An unusual hamartoma of the pigment epithelium and retina simulating choroidal melanoma and retinoblastoma. Trans Am Ophthalmol. 1973;71:171-83. 2. Arepalli S, Pellegrini M, Ferenczy SR, Shields CL. Combined hamartoma of the retina and retinal pigment epithelium: findings on enhanced depth imaging optical coherence tomography in eight eyes. Retina. 2014;34(11):2202-07. 3. Shields JA, Shields CL. Combined hamartoma of the retina and retinal pigment epithelium. In: Shields JA, Shields CL, ed. Intraocular Tumors. An Atlas and Textbook. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:246-51. 4. Shields CL, Thangappan A, Hartzell K, et al. Combined hamartoma of the retina and retinal pigment epithelium in 77 consecutive patients: visual outcome based on macular versus extramacular tumor location. Ophthalmology. 2008;115(12):2246–52. 5. Dedania VS, Ozgonul C, Zacks DN, Besirli CG. Novel classification system for combined hamartoma of the retina and retinal pigment epithelium. Retina. 2018;38(1):12-9. 6. Arrigo A, Corbelli E, Aragona E, et al. Optical coherence tomography and optical coherence tomography angiography evaluation of combined hamartoma of the retina and retinal pigment epithelium. Retina. 2019;39(5):1009-15. 7. Kanski JJ, Bowling B. Kanski's clinical ophthalmology e-book: a systematic approach. Elsevier Health Sciences, 2015. 8. Kang HM, Koh HJ, Chung EJ. Spectral-domain optical coherence tomography of combined hamartoma of the retina and retinal pigment epithelium in neurofibromatosis. Korean J Ophthalmol. 2013;27(1):68-71. 9. Hartnett ME, Trese M, Capone A, et al. Pediatric retina: medical and surgical approaches. Philadelphia: Lippincott Williams and Wilkins; 2005:231-3. 10. Shields CL, Mashayeki A, Dai VV, et al. Optical coherence tomographic findings of combined hamartoma of the retina and retinal pigment epithelium in 11 patients. Arch Ophthalmol. 2005;123(12):1746-50. 11. Dedania VS, Ozgonul C, Zacks DN, Besirli CG. Novel classification system for combined hamartoma of the retina and retinal pigment epithelium. Retina. 2018;38(1):12-9. 12. Ting TD, McCuen BW II, Fekrat S. Combined hamartoma of the retina and retinal pigment epithelium: optical coherence tomography. Retina. 2002;22(1):98-101. 13. Yassin SA, Al-Tamimi ER. Familial bilateral combined hamartoma of retina and retinal pigment epithelium associated with neurofibromatosis 1. Saudi J Ophthalmol. 2012;26(2):229-234. 14. Martin K, Rossi V, Ferrucci S, Pian D. Retinal astrocytic hamartoma. Optometry. 2010;81(5):221-33. 15. Rowley, S. A., F. J. O'callaghan, and J. P. Osborne. "Ophthalmic manifestations of tuberous sclerosis: a population based study." British journal of ophthalmology 85.4 (2001): 420-423. 16. Shields CL, Benevides R, Maeterin MA, Shields JA. Optical coherence tomography of retinal astrocytic hamartoma in 15 cases. Ophthalmology. 2016;113(9):1553-7. 17. Bagheri N, Wajda B, Calvo C, Durrani A. The Wills eye manual: Office and emergency room diagnosis and treatment of eye disease. Lippincott Williams & Wilkins, 2016. 18. Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509-16. 19. Tugal-Tutkun I. Pediatric uveitis. J Ophthalmic Vis Res. 2011;6(4): 259-69. 20. Donaldson MJ, Pulido JS, Herman DC, et al. Pars planitis: a 20-year study of incidence, clinical features and outcomes. Am J Ophthalmol. 2007;144(6):812-7. 21. Ozdal PC, Berker N, Tugal-Tutkun I. Pars planitis: epidemiology, clinical characteristics, management and visual prognosis. J Ophthalmic Vis Res. 2015;10(4):469-80. 22. Pendergrass TW, Davis S. Incidence of retinoblastoma in the United States." Arch Ophthalmol. 1980; 98(7):1204-10. 23. Scupola A, Grimaldi G, Sammarco MG, et al. Multimodal imaging evaluation of combined hamartoma of the retina and retinal pigment epithelium. Eur J Ophthalmol. 2020;30(3):595-9. 24. Chawla, R, Kumar, V, Tripathy, K, et al. Combined hamartoma of the retina and retinal pigment epithelium: an optical coherence tomography-based reappraisal. Am J Ophthalmol. 2017;181:88-96. 25. Schachat AP, Shields JA, Fine SL, et al. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology. 1984;91(12):1609-14. 26. Shields JA, Shields CL. Tumors and related lesions of the pigmented epithelium. Asia Pac J Ophthalmol (Phila). 20176(2):215-23. 27. Mason JO III, Kleiner R. Combined hamartomas of the retina and retinal pigment epithelium associated with epiretinal membrane and macular hole. Retina 1997;17:160–162. 28. Kaye LD, Rothner AD, Beauchamp GR, et al. Ocular findings associated with neurofibromatosis type II. Ophthalmology 1992;99:1424–1429. 29. Asthagiri, Ashok R., et al. "Neurofibromatosis type 2." The Lancet 373.9679 (2009): 1974-1986. 30. Rouleau, Guy A., et al. "Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2." Nature 363.6429 (1993): 515-521. 31. Rouleau, Guy A., et al. "Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22." Nature 329.6136 (1987): 246-248. 32. Rouleau, Guy A., et al. "Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2." Nature 363.6429 (1993): 515-521. 33. Lee, Joo Yong, et al. "Merlin, a tumor suppressor, interacts with transactivation-responsive RNA-binding protein and inhibits its oncogenic activity." Journal of Biological Chemistry 279.29 (2004): 30265-30273. 34. Ardern-Holmes, Simone, Gemma Fisher, and Kathryn North. "Neurofibromatosis type 2: presentation, major complications, and management, with a focus on the pediatric age group." Journal of child neurology 32.1 (2017): 9-22. 35. Ruggieri, Martino, Andrea Domenico Praticò, and Dafydd Gareth Evans. "Diagnosis, management, and new therapeutic options in childhood neurofibromatosis type 2 and related forms." Seminars in pediatric neurology. Vol. 22. No. 4. WB Saunders, 2015. 36. Evans, DG, Huson, SM, Donnai, D. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841–846. 37. Evans, D. Gareth R. "Neurofibromatosis type 2 (NF2): a clinical and molecular review." Orphanet journal of rare diseases 4.1 (2009): 1-11. 38. Evans, D. Gareth R., et al. "Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought." Otology & Neurotology 26.1 (2005): 93-97. 39. Lalwani A. Neurofibromatosis type 2. In: Current Diagnosis & Treatment in Otolaryngology - Head & Neck Surgery. The McGraw-Hill Companies, Inc; 2004: Chapter 62. 40. Evans, D. Gareth R., et al. "A clinical study of type 2 neurofibromatosis." QJM: An International Journal of Medicine 84.1 (1992): 603-618. 41. Evans, D. Gareth R., M. Sainio, and Michael E. Baser. "Neurofibromatosis type 2." Journal of medical genetics 37.12 (2000): 897-904. 42. Bosch, Martina M., et al. "Ophthalmologic findings and long-term course in patients with neurofibromatosis type 2." American journal of ophthalmology 141.6 (2006): 1068-1077. 43. Ragge, Nicola K., et al. "The ocular presentation of neurofibromatosis 2." Eye 11.1 (1997): 12-18 44. Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 1994;52:450–461. 45. Mautner, Victor-Felix, et al. "The neuroimaging and clinical spectrum of neurofibromatosis 2." Neurosurgery 38.5 (1996): 880-886. 46. Ragge, Nicola K., et al. "Ocular abnormalities in neurofibromatosis 2." American journal of ophthalmology 120.5 (1995): 634-641 47. Parry DM, MacCollin MM, Kaiser-Kupfer MI, et al. Germline mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet 1996;59:529–539. 48. Lee HBH, Garrity JA, Cameron JD, et al. Primary optic nerve sheath meningioma in children. Surv Ophthalmol. 2008;53(6):543-58. 49. Baser ME, Friedman JM, Aeschliman D, et al. Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet 2002;71:715–723. 50. Grant EA, Trzupek KM, Reis J, et al. Combined retinal hamartomas leading to the diagnosis of neurofibromatosis type 2. Ophthalmic Genet. 2008;29(3):133-8. |
