There may be times a clinician sees a pattern of dots, spots, lines or a combination of all three on both corneas and wonders if the patient has a corneal dystrophy or a corneal degeneration. Corneal dystrophies are inherited conditions—usually passed on as autosomal dominant traits—characterized by a specific bilateral, often symmetric pattern of opacities. These opacities are initially found in the central cornea of younger individuals, and over time become denser and spread to the periphery. Some opacities are small and do not affect vision early on but coalesce as time progresses, resulting in reduced visual function. Most are slowly progressive, leaving the patient asymptomatic for years.
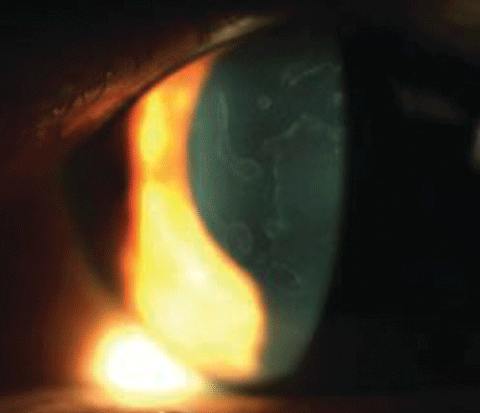 |
| Map-type changes in epithelial basement membrane dystrophy. |
Dystrophies are rarely associated with systemic disease and are not the result of inflammation. Therefore, any unusual pattern of corneal opacities associated with corneal neovascularization is not a dystrophy and clinicians should investigate other etiologies. Most dystrophies do not require any surgical intervention until later in life, and advances in surgical procedures have resulted in faster recovery of visual function.
Since corneal dystrophies are most often autosomal dominant in inheritance, clinicians should examine family members, given as many as 50% can be affected as well. Corneal dystrophies should not be confused with corneal degenerations, which tend to be asymmetric opacities in the periphery and are the result of aging and metabolic changes—as is the case with crocodile shagreen, Vogt’s girdle and arcus senilis—and inflammation, as seen in band keratopathy and Salzmann’s nodular degeneration.
Categorizing Corneal Dystrophies
Corneal dystrophies are categorized by the layers in which the opacities are found. However, new discoveries in the mutations that lead to the corneal dystrophies have increased our knowledge of their pathophysiology and may one day result in reclassification. Currently there are a number of dystrophies that have different phenotypic variations but are the result of mutations that occur in the same gene. Knowing the mutations that cause the dystrophies we see clinically will increase our understanding of the pathophysiological pathways that result in the deposition of the abnormal proteins interfering with corneal function and vision. This will someday lead to the development of medications that can interfere with these pathways to reduce deposition and the subsequent need for treatment.
Superficial corneal dystrophies affect the corneal epithelium, Bowman’s layer and, initially, the anterior portion of the stroma. Some will also result in changes that affect the epithelial layers of the cornea. These more superficial dystrophies are amenable to treatment with less invasive surgical procedures than a full penetrating keratoplasty (PK) or deep anterior lamellar keratoplasty (DALK). Recognizing a corneal dystrophy is important for prognosis of vision loss and early treatment to help avoid future vision loss.
The goal of the International Committee for Classification of the Corneal Dystrophies, formed in 2005, was to devise a new classification system for the corneal dystrophies. The committee divided the dystrophies into four categories, published in 2008, depending upon the phenotypic presentation that defined the condition and the genetics, such as whether the identification of the mutation was known, it was mapped to a specific location on a gene or the gene was not yet identified:1
Category 1: A well-defined corneal dystrophy in which the gene is mapped and the mutation that causes the dystrophy is known.
Category 2: A well-defined corneal dystrophy that is mapped to one or more specific chromosomal loci but the causative gene or genes are not known.
Category 3: A well-defined corneal dystrophy that has not been mapped to any specific chromosomal locus.
Category 4: A suspected or new corneal dystrophy that has not been well-defined as a corneal dystrophy.
As more genetic information is acquired, category 2, 3 and 4 dystrophies will ultimately become category 1 dystrophies.
Detecting Opacities: Biomicroscopy and OCT
The ability to detect corneal dystrophies is dependent on good biomicroscopic technique and various methods of illumination, as some of the early changes may be subtle. Direct illumination using a broad, oblique beam allows clinicians to identify the number, types and location (central/peripheral) of the opacities. Narrowing the beam to an optic section will reveal the corneal layer that is affected. Indirect illumination using sclerotic scatter and retroillumination through a dilated pupil will uncover more opacities that may not be evident with direct illumination. Newer technologies such as anterior segment optical coherence tomography (OCT) allow for more precise identification of the affected layers than biomicroscopy.
Treatment
Many superficial corneal dystrophies affect the integrity of the corneal epithelium, resulting in recurrent corneal erosions (RCEs). Over time, the RCEs as well as the dystrophy itself result in an irregular epithelial surface, which can affect vision. In addition, the RCEs themselves cause pain and photophobia. Treatment is always case specific, but it is initially aimed at treating the RCEs to reduce the pain and photophobia. Eventually other treatment modalities will be needed, such as specialty fit contact lenses for the irregular astigmatism caused by the RCEs and surgical procedures such as superficial keratectomy (SK), lamellar keratectomy (LK) and phototherapeutic keratectomy (PTK).
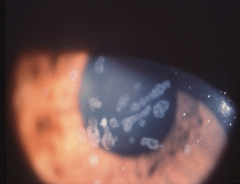 |
| Granular dystrophy in the patient’s 45-year-old mother with 20/40 visual acuity. |
These procedures are minimally invasive compared to PK, which results in longer recovery times. PTK uses an excimer laser (193nm) to remove the affected superficial layers of the cornea by photoablation.2 One pulse of laser can remove 0.25µm of corneal tissue, so it is quite precise and results in a smooth and regular corneal surface, which improves visual outcomes.2 It is used regularly to treat superficial corneal dystrophies affecting the epithelial and stromal layers for relief of symptoms secondary to RCEs and for visual improvement by removing or reducing the superficial corneal deposits associated with the corneal dystrophy. PTK should not be confused with photorefractive keratectomy (PRK), which is used to reduce refractive error. But because corneal dystrophies affect the central cornea, the ablation procedure mimics that of myopia correction. The result is an induced hyperopia, the amount of which is dependent on the ablation depth. This may be countered to some degree by simultaneously performing an anti-hyperopia treatment with the laser. Although PTK may induce refractive error, the patient’s best-corrected visual acuity and comfort may improve.
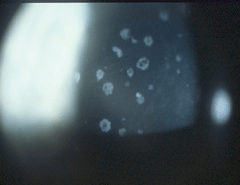 |
| Granular dystrophy in a 22-year-old asymptomatic female with 20/20 visual acuity in both eyes. |
If clinicians suspect a corneal dystrophy, they should initially try to examine family members and, if in doubt, refer to a corneal specialist for confirmation based on the phenotypic presentation. Since many dystrophies progress slowly, the patient can be monitored. Anterior segment photo documentation with OCT is important when the patient is being followed. Genetic confirmation is indicated if a family member is interested or if the clinical picture is not diagnostic. Testing for the transforming growth factor beta-induced (TGFBI) gene—a common gene that has mutations causing some of the corneal dystrophies—is available commercially, and insurance may or may not reimburse the patient. Clinicians should educate patients about what to expect regarding deterioration of vision with increasing opacification, symptoms of RCEs, if that is a part of the dystrophy, and potential treatment options if and when necessary.
Epithelial Dystrophies
A number of corneal dystrophies affect the cornea’s epithelial layer:
• Epithelial basement membrane dystrophy (EBMD). The most common epithelial dystrophy is EBMD, previously referred to as map-dot-fingerprint dystrophy. As its name suggests, this dystrophy is characterized by patterns that look like small continents on a map, fingerprint swirls, small dot opacities and bleb or milky-white opacities. These patterns are created by the opacities composed of abnormal basement membrane material. The deposits can occur within the epithelial layers (maps), between the epithelial cells (fingerprint swirls) or within the epithelial cells (dots). Some of these changes can best be appreciated after instillation of fluorescein dye, since wetting is irregular over the maps and fingerprint opacities.
RCEs can occur after the third decade of life. Patients presenting with RCE with no prior history of corneal trauma, surgery or contact lens wear should be suspect for an epithelial dystrophy, although corneal dystrophies affecting other layers may also result in RCEs as well.
EBMD is not always a true dystrophy, however, in the sense that some of these changes can also appear following corneal trauma or surgery. Some forms of EBMD have been noted to be of autosomal dominant inheritance. Mutations are localized to the TGFBI gene on chromosome 5, making some forms of EBMD a category 1 dystrophy.3 The TGFBI gene, also known as the keratoepithelin gene, interacts with collagen and is responsible for corneal development and healing. Mutations in TGFBI actually cause seven corneal dystrophies in total, with varying phenotypic expression, affecting the epithelium, Bowman’s membrane as well as the stroma.
The RCEs can be treated with antibiotics, artificial tear solutions and bandage contact lenses. Repeated RCEs that cause corneal irregularity and reduced vision can be successfully treated with anterior stromal puncture, epithelial debridement and polishing as well as PTK.4
• Meesmann dystrophy. This dystrophy is characterized by diffuse, tiny, gray-white vesicles that extend to the limbus.5 Unlike most other corneal dystrophies, Meesmann dystrophy is often first apparent on exam at quite an early age. It progresses slowly until such time as there are RCEs, which occur when the vesicles burst. Meesmann dystrophy usually causes no effects on vision until middle age.
The mutation that causes Meesmann dystrophy has been localized to two keratin-specific genes, KRT3 on chromosome 12q12 and KRT12 on chromosome 17q12, making Meesmann dystrophy a category 1 corneal dystrophy.6 Treatment is initially targeted to treat the pain and photosensitivity of RCEs. Eventually PTK and LK may be needed to treat the irregularity of the corneal surface due to repeated RCEs.7
• Lisch corneal dystrophy. Originally named whirled, band-shaped, microcystic dystrophy after the patterns of interesting opacities, this dystrophy has since been shortened to Lisch corneal dystrophy after the ophthalmologist Karl Lisch who first described it.8 It is a rare dystrophy inherited as a sex-linked trait and is characterized by interesting, almost artistic patterns of whirls, feathery-shaped and linear opacities, all consisting of small deposits in the epithelial cells. On histologic exam, these deposits are due to the vacuolization of the epithelial cell cytoplasm and disappear when patients wear gas permeable contact lenses.
Because the clinical appearance of tiny vacuoles is similar to Meesmann corneal dystrophy, the question arose whether Lisch dystrophy was a variant of Meesmann dystrophy. However, the gene that causes Lisch dystrophy has been mapped to the X chromosome, Xp22.3, unlike Meesmann dystrophy, but the mutation has not yet been identified, making it a category 2 dystrophy.9 This is a perfect example of how genetics has helped differentiate a dystrophy that is phenotypically similar to another.
• Epithelial recurrent corneal erosion dystrophy (ERED). Perhaps one of the worst of the epithelial dystrophies is ERED, as it results in RCEs by age five.10 Patients are besieged by repeated attacks of RCEs that can be triggered by smoke, dry air, upper respiratory infection and minimal trauma. Treatment is problematic, and attacks can last weeks. Eventually, the cornea scars from repeated RCEs and small keloids form, resulting in irregular corneas and vision loss. While PTK has been attempted, this dystrophy is difficult to treat. By the third to fourth decade of life, patients’ RCEs subside, but the corneal damage has already occurred. A corneal graft may be needed in about 25% of cases.10
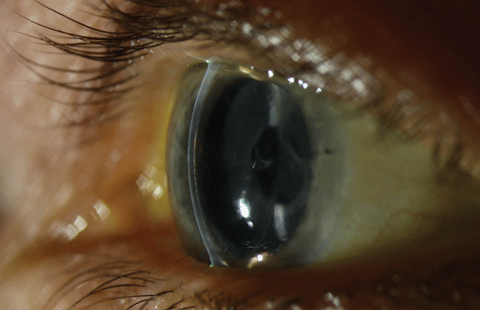 |
| Posterior corneal changes in a patient with EBMD and Fuch’s dystrophy. Photo: Christine W. Sindt, OD. Click image to enlarge. |
Bowman’s Layer Dystrophies
There are a few dystrophies of Bowman’s layer, but the most common is Reis-Buckler corneal dystrophy (RBCD). This dystrophy is characterized by ring-shaped opacities that result from localized areas of Bowman’s membrane thickening.11 It results in epithelial irregularity that causes irregular astigmatism and RCEs. Visual acuity is usually good until the later decades when the ring-shaped opacities become more pronounced, spreading to the periphery.Treatment for the RCEs and corneal irregularity by therapeutic contact lenses, superficial keratectomy (SK), PTK and PK have all been used to treat RBCD; however, the dystrophy recurs within a few years. Investigators report that mitomycin C can help prevent recurrences.12 The mutation that causes RBCD has been identified in the TGFBI gene on chromosome 5q31, making it a category 1 dystrophy.13
As RBCD progresses, the ring-shaped opacities spread to the corneal periphery. In addition, the RCEs eventually cause scarring.
 |
| Advanced granular dystrophy in a 79-year-old male with 20/200 visual acuity. |
Thiel-Benkhe dystrophy (TBCD), which also affects Bowman’s layer, is less progressive than Reis-Buckler dystrophy, but has the same opacities. Some consider TBCD to be a variant of RBCD. Research also describes another similar dystrophy in only one family, which may also simply be a variant of RBCD.14
Anterior Stromal Dystrophies
There are myriad corneal dystrophies that affect the stroma, since the stroma is the thickest layer of the cornea. But only stromal dystrophies that affect the anterior stroma are most amenable to treatment with PTK. Many of these dystrophies also cause RCEs and have an effect on epithelial function and structure. Although PTK has been attempted in some of the other corneal dystrophies that affect the deeper stroma, PTK has not been successful.
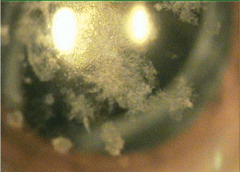
• Granular dystrophy. One of the most common of the anterior stromal dystrophies is granular dystrophy. As its name suggests, granular dystrophy is characterized by powdery and granular or crumb-like opacities in the central cornea that are composed of hyaline.15The stroma between these opacities is clear, and vision is normal for most of the patient’s life. Vision slowly deteriorates, but treatment is not usually necessary until the seventh or eighth decade of life.
The mutation that causes granular dystrophy is on the TGFBI gene on chromosome 5q31, one of the seven corneal dystrophies resulting from TGFBI mutations. The mutation results in accumulation of hyaline, causing the types of deposits seen, which can be treated with PTK.16
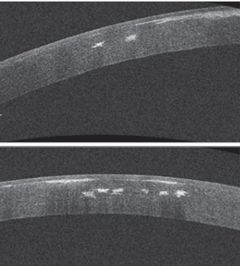 |
| OCT of the patient’s cornea demonstrates the anterior and mid-stromal nature of these deposits. |
• Lattice dystrophy. Another common stromal dystrophy which may be treated with PTK is lattice dystrophy. Lattice dystrophy gets it name from the lattice-like network of refractile lines that lie in the anterior stroma. There are several types of lattice dystrophy, all of which are characterized by the same refractile lattice-type lines. In all types, the lattice lines are made up of amyloid.17 Type 1 lattice dystrophy is a typical dystrophy, inherited as an autosomal dominant trait. The lines start in the central cornea and spread to the periphery in the later years. Because the stroma between is relatively clear early on, vision is fairly good until later in life when the lattice lines spread to the periphery of the cornea and amyloid plaques form. Lattice dystrophy may also cause RCEs, which may be treated by PTK. Such treatment may be effective early on, but some experts feel that the UV light in PTK may increase formation of the deposits. The mutations that cause type 1 lattice dystrophy are also in TGFBI in chromosome 5q31.
Type 2 lattice dystrophy manifests the same refractile lattice lines as type 1, but is one of the rare dystrophies associated with a systemic disorder, namely systemic amyloidosis—a disorder of amyloid metabolism caused by mutations on the gelsolin gene on chromosome 6.18 Amyloid will deposit in the skin, sclera and peripheral nerves as well as the cornea. The lattice deposits seen in type 2 develop later in life than in type 1 and are thicker and more peripheral. This type does not cause RCEs.
A number of other types of lattice dystrophy have been reported in the literature depending on the pedigree, mode of inheritance and phenotypic expression. These are typically later-onset dystrophies with deeper stromal opacities with various forms of amyloid deposition.19,20
• Combined granular-lattice dystrophy (Avellino corneal dystrophy). A pedigree was reported in the small town of Avellino, Italy, that appeared to have corneal deposits described as being both granular and lattice-like refractile lines. The granular opacities predominated in the younger individuals, while the lattice lines appeared in the older individuals as the granular opacities coalesced and thickened.21 Histology demonstrated that in this combined granular-lattice dystrophy, the granular opacities are composed of hyaline, and the lattice lines are composed of amyloid. Since it was first described, the name has been changed from Avellino corneal dystrophy to combined granular-lattice dystrophy. Since both dystrophies are the result of mutations in the TGFBI gene, it is possible that the mutations segregate together.
Advanced lesions have been treated with PTK in combined dystrophy, but research shows PTK can result in more rapid and advanced recurrence of the opacities, presumably due to the ultraviolet light.22
• Crystalline dystrophy of Schnyder. Crystalline dystrophy, as its name suggests, is a stromal dystrophy characterized by crystalline, refractile deposits in the stroma. These deposits are composed of cholesterol and phospholipids.23 Early cases present with central crystalline deposits with minimal effect on visual acuity. With time, some patients may also have an associated arcus composed of cholesterol.
Although corneal dystrophies are rarely associated with a systemic disorder, a young patient presenting with crystalline deposits should be worked up for systemic dyslipidemia. Mutations in the UBIAD1 gene, which may regulate or play a role in cholesterol biochemistry, transport or storage, have been reported in some pedigrees.24
Corneal Dystrophy Discoveries
Recently, the corneal dystrophies have undergone an evolution of categorization and genetic discovery. Many patients suffer from pain and photophobia secondary to the RCEs that are associated with many of the superficial corneal dystrophies, as well as visual compromise due to the increased density of the opacities with age and the corneal irregularity that results from repeated RCEs.
When specialty contact lenses and topical drops are no longer effective, PTK is the most commonly used treatment for smoothing the irregular corneal surface and removing or reducing the density of the opacities associated with the superficial corneal dystrophies; however, success varies and recurrences are common.
The identification of the mutation responsible for the corneal dystrophy is a critical step toward understanding the pathophysiology of the formation of the deposits. Once the pathways that form the abnormal proteins associated with the corneal dystrophies are identified, other treatments may be developed to interfere with the production of these abnormal proteins. This ultimately results in less deposition of opacities, less symptoms and less need for surgical intervention.
Early diagnosis will be even more important for timely intervention once new treatment becomes available. In the meantime, patients have access to therapeutic options that offer better comfort and vision.
Dr. Bass is a distinguished teaching professor at the SUNY State College of Optometry. She lectures on hereditary diseases of the eye.
|
1. Weiss J, Maler H, Lisch W, et al. The IC3D classification of the corneal dystrophies. Cornea. 2008; 27(Suppl 2):S1–83. |
