Retinal Vascular Miniseries |
| This article is the last in a four-part series about retinal vasculature. Follow the links below to read the other installments: Part 1: Recognizing Abnormal Vasculature Part 2: Read the Retinal Vasculature Like a Pro Part 3: Imaging Motion: a Review of OCT-A |
Ocular vascular tumors are rare, and you may have few opportunities to diagnose one. However, when present, they may be the initial finding of one or more serious systemic deficits, which will require you to mobilize and perhaps even lead a multidisciplinary team to provide collaborative patient care.
In the posterior segment of the eye, tumors are classified by their location: retinal or choroidal. Retinal vascular tumors are categorized into one of four main types; retinal capillary hemangiomas, retinal vasoproliferative tumors, cavernous hemangiomas and racemose hemangiomatosis (Wyburn-Mason syndrome). Choroidal vascular tumors are categorized as either circumscribed or diffuse.
This article, last in our four-part miniseries on the retinal vasculature, reviews how to clinically distinguish these tumors and discusses their associations.
Retinal Capillary Hemangiomas
Retinal capillary hemangiomas (RCH) are highly vascularized tumors that typically present as a red, round, circumscribed mass, more commonly in the midperiphery (85%) or less commonly juxtapapillary (15%).1,2 When peripheral, they are often in the superotemporal or inferotemporal quadrants and can easily be found by following their feeder vessel extending from the optic nerve.1 The majority of RCHs are solitary (66%) but they can also present as multiple lesions (33%).2-4 They demonstrate no sex, racial or laterality predilection.1
While RCHs are most commonly endophytic in presentation and portray the classic round mass in the inner retina, they may also be exophytic or sessile, varying in appearance.3,4 Juxtapapillary exophytic lesions are sometimes misdiagnosed as optic disc edema as they may blur the disc margin.4 Most RCHs are asymptomatic and are, therefore, commonly diagnosed incidentally between the ages of one and 40 years, with a mean age of 25.1-3 However, their secondary effects may elicit symptoms. Secondary complications include intraretinal and subretinal exudation in the vicinity of the hemangioma, uncommonly a macular star (a circular distribution of exudates around the macula) and rarely retinal or vitreous hemorrhage (less than 3%).3 Advanced cases may present with tractional or exudative retinal detachment.1,3,4 Symptoms in these instances may include loss of vision, flashes, floaters, photopsias and metamorphopsias.1-3
Although patients can have RCHs alone, RCHs are also associated with Von Hippel-Lindau (VHL) disease.3,4 RCHs are the most common, and often earliest, presentation of VHL.3-5 Twenty to 58% of patients with RCHs have VHL disease as the underlying cause.3 VHL is an inherited autosomal dominant syndrome that has age-dependent penetrance.3,5 The onset is classically between the first to fourth decade of life.3 VHL manifests in a range of benign and malignant tumors, including hemangiomas of the retina and central nervous system (CNS), renal cell carcinoma, pheochromocytomas, pancreatic carcinoma and cysts in the liver, kidneys and pancreas.2,5
VHL is caused by a mutation in a tumor suppressor gene called VHL gene, resulting in the upregulation of angiogenic factors, including vascular endothelial growth factor (VEGF). One study found an increased incidence of retinal neovascularization similar to that found in diabetic retinopathy.6 VHL gene may also present with additional rare retinal manifestations that include atypical retinal vascular hamartomas and twin blood vessels, paired artery and veins that are separated by less than one venual width for the extent of one disc diameter.3,7,8 Associated CNS hemangiomas may involve the visual pathway, resulting in optic nerve compression and chiasmal syndrome.3
The average life expectancy of patients with VHL is less than 50 years, with morbidity most commonly due to renal cell carcinoma.1,3,5 Early detection of affected gene carriers can play a critical role in improved prognosis. Today, commercially available genetic testing for VHL yields exceptionally high detection rates (99%). Therefore, this testing is indicated in all patients with RCHs.2 Primary eye care providers may have a vital role in the early screening and diagnosis of VHL in relatives of those already diagnosed.5,9
While RCH is largely a clinical diagnosis, ancillary testing can help monitor regression, growth or the production of intraretinal or subretinal fluid.1,2 Due to the vascular nature of RCHs, fluorescein angiography (FA) tends to be the most revealing test.1,4 On FA, the tumor displays fine capillary filling that rapidly becomes homogenous and demonstrates late leakage.3,4 While OCT does not contribute to the diagnosis of RCHs, it can help monitor success of treatment, such as tracking resolution of subretinal fluid.2 On A-scan ultrasonography, the initial spike is followed by high internal reflectivity due to blood flow, and on B-scan there is a well-demarcated retinal lesion without choroidal involvement.3 While thin orbital sections on magnetic resonance imaging (MRI) can detect retinal capillary hemangiomas greater than 2mm thick, MRI is most beneficial for detecting associated CNS hemangiomas using gadolinium enhancement.3 In addition, enhanced CT scanning can detect other organ involvement.3
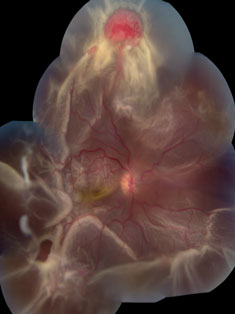 | 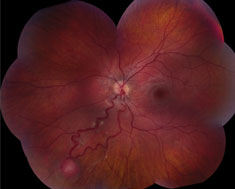 |
| At left, extensive circumferential retinal detachment is secondary to a retinal capillary hemagioma in Von Hippel-Lindau disease. Above, peripheral retinal capillary hemangioma is located inferior-nasally with dilated, tortuous vasculature in Von Hippel-Lindau disease. Click images to enlarge. |
Treatment for RCH is dependent on size, location and associated findings such as exudation (25%) or retinal detachment (16%).1,3,4 If the tumor is less than 500µm without exudation or subretinal fluid, the lesion may simply be observed.1,2,4 However, if the patient’s visual acuity becomes affected or threatened, anti-VEGF injections, laser photocoagulation, cryotherapy, photodynamic therapy (PDT), radiotherapy or other vitreoretinal procedures may be indicated.1,2,4
Similarly, visual prognosis is dependent on number, size, location and degree of exudation or traction.2 Severe visual impairment in affected eyes is more likely with increased age, juxtapapillary location of lesion and increased number of peripheral lesions.1 Overall, there is a guarded prognosis with more than 25% of eyes developing permanent vision loss and 20% having a visual acuity worse than 20/100 in at least one eye.1,2
Racemose Hemangiomatosis
This congenital form of arterial-venous malformation (AVM) classically presents as dilated tortuous retinal vessels spanning from the optic disc to the periphery.2 The clinical appearance is categorized into three groups:1,2,7
- Group I has an abnormal capillary network
- Group II lacks any capillary network
- Group III presents with dilated tortuous vessels that make it difficult to differentiate arteries and veins.1,2,10
Distinguishing features of AVMs include the absence of one or more feeder vessels and the absence of exudation or subretinal fluid.1 Patients are typically asymptomatic and the malformations are usually incidental findings.1 Some visual disturbances may occur depending on the size and location of the retinal AVM. Additionally, vessel occlusions may result due to turbulent blood flow, damage to vessel walls and thrombosis formation.10 Retinal vessel occlusions can cause direct vision loss themselves or secondary vision loss from macular edema or if neovascular glaucoma (NVG) develops.2,10
Retinal AVMs can be associated with the rare phakomatosis, Wyburn-Mason syndrome (WMS), also known as congenital unilateral retinocephalic vascular malformation syndrome.10 Phakomatoses, more descriptively known as neuro-oculocutaneous syndromes, are a group of conditions that result in hamartomas, which are benign tumors that arise from normally existing tissue. WMS, however, is not commonly characterized by cutaneous involvement as with other phakomatoses.2
WMS is a congenital, non-hereditary sporadic disorder with no sex or ethnic predilection.1,2 The syndrome may involve other tissues including skin, bones, kidneys, muscles, GI and the brain, resulting in a variety of systemic symptoms.2 Cerebral vascular changes in the brainstem and subarachnoid hemorrhage may elicit nystagmus or affect ocular motility, causing diplopia.10 Uncommonly, there may be optic disc atrophy or edema and visual field defects due to cerebral AVMs affecting the visual pathway.10 Unilateral proptosis is indicative of orbital involvement and is secondary to AVMs involving the orbit or intracranial vessels.10
These patients may also present with dilated conjunctival vessels, similar to the presentation of a cavernous-carotid fistula.10 The most critical systemic manifestation occurs when intracranial AVMs hemorrhage, which may result in mortality.2 While the incidence of co-existing intracranial AVMs with retinal AVMs is 30%, only 8% of patients with intracranial AVMs have retinal AVMs.2 The current recommendation is for all patients with retinal AVMs to undergo brain and orbital neuroimaging to rule out cerebral AVMs.10
Due to their distinct clinical appearance, retinal AVMs are a clinical diagnosis; however, for confirmation and staging of the condition, fluorescein angiography demonstrates the anomalous arteriovenous communication and the lack or presence of capillaries.2 Of note, there will be no leakage on FA and, on OCT, there will be no subretinal fluid.1 In group III retinal AVMs, FA cannot distinguish arteries from veins. For detecting associated intracranial AVMs, MRI is currently the imaging of choice to determine the size and extent of the AVM as well as its impact on surrounding neurogical tissue. On the other hand, conventional angiography is still the standard for characterizing the AVM due to its ability to visualize the angio-architecture of the lesion.2,11,12
Retinal AVMs are typically not amenable to treatment.1 Treatment is, therefore, targeted towards resulting comorbidities such as retinal ischemia and NVG.1,2 Clinicians can consider several treatment strategies, including anti-VEGF injection, panretinal photocoagulation, glaucoma surgery or glaucoma medication.1,2,10 The prognosis is guarded, as there is high risk for functional vision loss due to retinal ischemia or NVG.1,2,10 Unlike brain AVMs, retinal AVMs rarely hemorrhage.2
Cluster of Grapes
Retina cavernous hemangionas are rare benign, thin-walled intraretinal vascular tumors.7,13 They manifest as multiple venous aneurysms along one or more veins, and less often appear on the optic nerve head.13 From their configuration and purple color, their appearance is commonly described as a “cluster” or “bunch of grapes in the retina.”9 These lesions tend to be unilateral and solitary, and usually are non-progressive.13 Their sizes are variable, and the larger ones are at a higher risk of leading to complications such as vitreous hemorrhages, preretinal fibrosis and traction, hyphema and secondary glaucoma.13
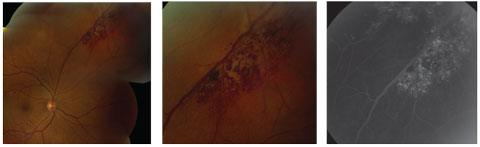 |
| This cavernous hemangioma demonstrates the classic “cluster of grapes” distribution. At right, the corresponding fluorescein angiography demonstrates the persistent hyperfluorescence in the late phase. Click image to enlarge. |
Retinal cavernous hemangiomas are usually isolated; however, they can be autosomal dominant, which can have associated central nervous system, hepatic and cutaneous vascular anomalies. In these instances, they are considered phakomatoses.14-16 Investigators estimate that about 5% of retinal cavernous hemangiomas have associated cerebral cavernous malformations.14 There is a strong expression of cerebral cavernous malformation genes in both the brain and the retina, which code for proteins important in endothelial cell structure and function.14
Fluorescein angiography is beneficial in clinching the diagnosis. Early hypofluorescence is seen in the vascular filling phase with persistent hyperfluorescence in the recirculation phase due to the slow-flow nature of the lesions.13,16 Stagnant erythrocytes layer inferiorly in the saccules (hypofluorescent), leaving the plasma superiorly within each aneurysm (hyperfluorescent fluorescein capping).13-16 In addition, due to an intact endothelium, there is no extravascular leakage, which is a key differentiator from retinal telangiectasias.13-16
These lesions possess a good visual prognosis.17 Testing should be done to rule out similarly presenting lesions, including capillary hemangiomas, Coats’ disease and Leber’s miliary aneurysms.14 If there are recurrent vitreous hemorrhages, particularly from larger lesions, possible treatments include focal photocoagulation, cryotherapy, vitrectomy or diathermy in an effort to sclerose leaky tumor vessels.17 With treatment, however, there is an increased risk of vitreous hemorrhage as well as secondary tractional membrane and retinal detachment.17
Tomato-Ketchup Fundus
Choroidal hemangionas are rare hamartomas that are highly vascular, yet benign in nature.1,18 They are composed of vascular channels lined with endothelium.1 Despite their intrinsic vascularity, they lack any feeder vessels but can have nearby dilated choroidal vasculature.1,19 More than 90% of the time they are found in Caucasians, but do not have a sex predilection.18,19 They range in shape from round to oval; in color, from orange-red to pink to yellow to amelanotic; and are variable in size.1,18,19 They are typically unilateral solitary lesions that are nonproliferative; if they do enlarge, it is not due to cell proliferation but rather from venous congestion.18,19 Choroidal hemangiomas are separated into two categories: circumscribed and diffuse.
Circumscribed choroidal hemangiomas, also known as discrete choroidal hemangiomas, represent about 50% of cases.11 While possibly congenital, these typically are not discovered until adulthood, since the symptoms are rare. If they are not congenital they arise later in life, between the third and sixth decades.1,18-20 Symptoms include blurred vision from a hyperopic shift, and metamorphopsia (if in the macula), photopsia and visual field defects secondary to serous retinal detachments and subsequent RPE and outer retinal degenerative changes.1,18,19 These tumors are mainly located posterior to the equator. They may often be found in the peripapillary region and adjacent to the macula.1,18,20
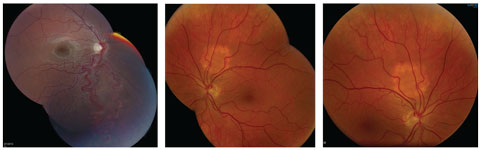 |
| At left, an engorged artery and vein in an arterial-venous malformation extends from the optic disc to the inferior peripheral retina in a patient with Wyburn-Mason syndrome. At middle and right are choroidal hemangiomas. Click image to enlarge. |
Despite their location, these lesions can often be missed due to indistinct margins and color that blends into the surrounding choroid.1,20 Specific features will help with lesion identification, such as the appearance of a brown ring around the tumor margin secondary to tumor compression of the surrounding choroid, and yellow-white surface foci secondary to lipofuscin deposition from macrophage activity.19,20 Typically, these lesions average 7mm in diameter with a broad range of 3mm to 18mm, and 3mm in thickness with a range of 1mm to 7mm.1,20
Diffuse choroidal hemangiomas represent the other 50% of choroidal hemangiomas. Unlike circumscribed tumors, these have a syndromic association with Sturge-Weber.18 They are present at birth and involve a large portion of the choroid.1,20 They appear as a diffuse, orange choroidal thickening, creating the “tomato-ketchup fundus.”1 It is estimated that about 50% of Sturge-Weber syndrome patients have choroidal hemangiomas, which are mostly ipsilateral to their characteristic birthmark known as a “port-wine stain” or facial nevus flammeus.1,20
Fluorescein angiography may not be entirely revealing in these cases.18 ICG readily highlights the intrinsic vascular pattern within 30 seconds of injection (much earlier compared with other choroidal tumors).1,19 In the later stages, there is a “wash out” phenomenon, which represents loss of dye from the tumor.1 On B-scan there is a characteristic dome-shaped, acoustically solid lesion with regular structure and smooth contour. Upon A-scan, there is a high internal reflectivity between 50% and 100% from the multiple vascular channels but an absence of spontaneous vascular movements.1,18,19
Choroidal hemangiomas are typically observed as long as the patient is asymptomatic and the tumor location is not vision-threatening.20 However, given the similarity in appearance to amelanotic choroidal melanomas, aforementioned ancillary testing should be done to confirm the diagnosis.20
If symptoms do arise, treatment should be initiated to induce tumor atrophy and regression of subretinal fluid.1 Treatments include photocoagulation, photodynamic therapy, radiotherapy and transpupillary thermotherapy (TTT).1 While photocoagulation has been a staple, it is often ineffective at inducing tumor atrophy and has a 40% chance of failing to irradiate subretinal fluid.1,18 Both radiotherapy and TTT have demonstrated effectivness at tumor and fluid regression, but radiation increases the risk of optic neuropathy and radiation retinopathy with the added complications of cystoid macular edema, preretinal fibrosis formation and retinal vein occlusion.1,18
Dr. Brafman is an optometric resident at the Bascom Palmer Eye Institute.
Dr. Small is an optometric resident at Bascom Palmer Eye Institute.
Dr. Dunbar is the director of optometric services and the optometric residency supervisor at the University of Miami’s Bascom Palmer Eye Institute.
|
1. Singh AD, Rundle PA, Rennie IG. Retinal vascular tumors. In: Singh DA, Damato B, ed. Clinical Ophthalmic Oncology: Retinal Tumors. Berlin, Heidelberg: Springer Berlin Heidelberg, 2014:17-34. 2. Turell ME, Singh AD. Vascular tumors of the retina and choroid: diagnosis and treatment. Middle East Afr J Ophthalmol. 2010;17:191-200. 3. Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117-42. 4. Kase S, Ishida S. Retinal capillary hemangioma in von Hippel-Lindau disease: current concept, diagnosis and managements. J Trans Med Epidemiolol. 2014;2:1010. 5. Tootee A, Hasani- Ranjbar S. Von Hippel-Lindau disease: a new approach to an old problem. International J Endocrinol Metabol. 2012;10:619-24. 6. Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von hippel-lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371-8. 7. de Jong PT, Verkaart RJ, van de Vooren MJ, et al. Twin vessels in von Hippel-Lindau disease. Am J Ophthalmol. 1988;105:165-9. 8. Schmidt D, Neumann HH. Retinal vascular hamartoma in von hippel-lindau disease. Arch Ophthalmol. 1995;113:1163-7. 9. Hasani-Ranjbar S, Amoli MM, Ebrahim-Habibi A, et al. Mutation screening of VHL gene in a family with malignant bilateral pheochromocytoma: from isolated familial pheochromocytoma to von Hippel-Lindau disease. Familial Cancer. 2009;8:465-71. 10. Schmidt D, Pache M, Schumacher M. The congenital unilateral retinocephalic vascular malformation syndrome (Bonnet-Dechaume-Blanc syndrome or Wyburn-Mason syndrome): review of the literature. Surv Ophthalmol. 2008;53:227-49. 11. Luo CB, Lasjaunias P, Bhattacharya J. Craniofacial vascular malformations in Wyburn-Mason syndrome. J Chin Med Assoc. 2006;69:575-80. 12. Koenigsberg R. Brain imaging in arteriovenous malformation. October 28, 2015. http://emedicine.medscape.com/article/337220-overview-showall. Accessed September 25, 2016. 13. Shields JA, Eagle RCJ, Ewing MQ, et al. Retinal cavernous hemangioma: fifty-two years of clinical follow-up with clinicopathologic correlation. Retina. 2014;34:1253-7. 14. Reddy S, Gorin MB, McCannel TA, et al. Novel KRIT1/CCM1 mutation in a patient with retinal cavernous hemangioma and cerebral cavernous malformation. Graefe’s Arch Clin Exp Ophthalmol. 2010;248:1359-61. 15. Bloch E, Hakim J. Retinal cavernous hemangioma. Ophthalmology. 2015;122(10):2037. 16. Haug S. A 30-yr-old woman noted to have a lesion in her left eye. Case of the Month. West Coast Retina. December 2012. http://westcoastretina.com/dec-2012.html. Accessed September 5, 2016. 17. Alsulaiman SM, Abouammoh MA, Al-Dahmash SA, Abu El-Asrar AM. Is systemic infliximab therapy effective for retinal cavernous hemangioma? Saudi Medical Journal. 2014;35:1127-30. 18. Zablocki G, HC, Brady KD. Choroidal hemangioma: Central scotoma in the right eye. University of Iowa Health Care Ophthalmology and Visual Sciences. February 2, 2007. http://webeye.ophth.uiowa.edu/eyeforum/cases/65-Choroidal-Hemangioma-Photodynamic-Therapy.htm. Accessed September 9, 2016. 19. Karimi S, Nourinia R, Mashayekhi A. Circumscribed choroidal hemangioma. J Ophthal Vis Res. 2015;10:320-8. 20. Shields CL, Shields JA. Choroidal hemangioma. Seminars in Ophthalmology. 1993;8:257-64. |
