 A 41-year-old black female presented complaining of a wispy floater in the peripheral vision of her left eye. Onset was about two weeks earlier, but the symptoms have since disappeared.
A 41-year-old black female presented complaining of a wispy floater in the peripheral vision of her left eye. Onset was about two weeks earlier, but the symptoms have since disappeared.
The patient has been seen here since she was in her early 20s. She underwent LASIK approximately four years before this visit, with excellent results. We noted no significant medical history during her previous exams.
Uncorrected visual acuity was 20/20 O.U. Extraocular motility testing was normal. Confrontation visual fields were full to careful finger counting O.U., and her pupils were equally round and reactive without an afferent pupillary defect. The anterior segment exam was remarkable for very faint, circumferential corneal scars consistent with LASIK. IOP measured 13mm Hg O.U.
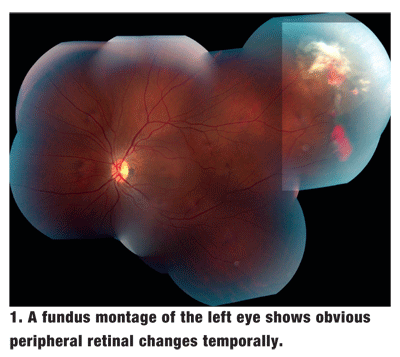
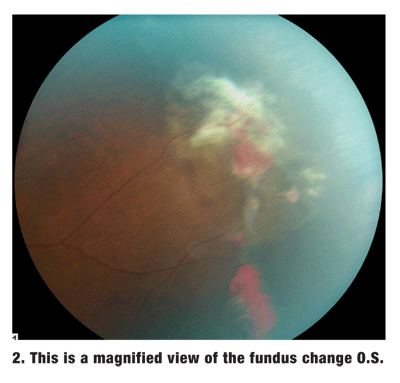
Dilated fundus exam of each eye showed a clear vitreous, a small cup with good rim coloration and perfusion, and a normal macula. The peripheral retina O.D. showed localized areas of nonspecific retinal pigment hyperplasia, and the peripheral retina O.S. showed the changes seen in figures 1 and 2.
Take the Retina Quiz
1. What do the changes in the peripheral retina represent?
a. Preretinal hemorrhage.
b. Retinal hole.
c. Neovascularization.
d. Capillary hemangioma of the retina.
2. What is the likely cause of the patients symptoms?
a. Symptomatic posterior vitreous detachment (PVD).
b. Vitreous hemorrhage.
c. Ophthalmic migraine.
d. Retinal detachment.
3. What is the correct diagnosis?
a. Nonspecific proliferative retinopathy.
b. Von Hippel-Lindau syndrome.
c. Sickle-cell retinopathy.
d. Vasoproliferative tumor.
4. How should this patient be managed?
a. Observation.
b. Laser photocoagulation.
c. Feeder vessel ablation.
d. Avastin (bevacizumab, Genentech).
For answers, see below.
Discussion
We expected our patient to have a normal eye exam. Her symptoms were vague, and her eye exams for the past several years were completely normal, except for myopia before she underwent LASIK. So, we were surprised to see these changes in the peripheral retina O.S. There is obvious fibrovascular proliferation and peripheral neovascularization.
These changes were indicative of proliferative sickle-cell retinopathy, even though her medical record indicated that she was healthy. I asked the patient if she had sickle-cell anemia, and she responded that she had sickle-cell trait (HbAS). Upon review of her medical record, we saw that this information was recorded many years earlier but never updated on subsequent exams.
Sickle-cell anemia (HbS) is a blood disorder that affects hemoglobin, the oxygen-carrying protein in all human erythrocytes. Sickle-cell anemia results from a genetic mutation that leads to structural abnormalities within the hemoglobin. Specifically, an abnormal globin protein chain causes the erythrocytes to lose some pliability. This, in turn, results in sickling of the erythrocytes and obstruction of the microcirculation. Because HbS is a multisystem disease, patients can develop chronic hemolytic anemia, episodes of recurrent pain and even organ dysfunction.
HbS develops when a person inherits an abnormal gene from each parent. If just one parent passes along the gene, the person has sickle-cell trait (HbAS).
Sickle-cell anemia is especially prevalent in blacks. An estimated 70,000 Americans have HbS, and approximately 2 million blacks in the
Other forms of HbS include hemoglobin S and hemoglobin C. The classification of HbS is based on the structural aberrations within the globin subunits or in the ability of hemoglobin to synthesize oxygen. The various forms of sickle-cell anemia include HbSS, HbCC, HbSC, and HbBeta () thalassemia, which can also be present with HbS (
Systemic disease is most common with HbSS, occurs less frequently in HbSThal and HbSC, and is exceedingly rare with sickle-cell trait. In contrast, HbSC and HbSThal are more likely to have retinal complications from sickle-cell disease.1 Our patient has sickle-cell trait.
People with sickle-cell trait are generally healthy and rarely develop systemic complications. They are more likely to develop ocular complications, but even these are unusual. Thats why we were surprised that our patient had ocular findings associated with her HbAS.
Myriad ocular changes can be seen in patients with sickle-cell anemia. The changes that occur in the anterior segment can be subtle and are often overlooked. These include conjunctival vasculature changes, in which the vessels have a comma or corkscrew shape. The conjunctival changes are more common in HbSS patients and are very rare in those with HbAS. Segmental iris atrophy can also develop.
The more significant changes occur within the posterior segment. These include salmon-patch hemorrhages, iridescent spots, black sunburst lesions, vascular occlusions and sea fan-shaped neovascularization. Sea fan neovascularization may result in vitreous hemorrhage and subsequent tractional and/or rhegmatogenous retinal detachment.
Proliferative retinopathy is present whenever sea fan neovascularization occurs. As with any other proliferative retinopathy, neovascularization occurs in response to localized ischemia. In sickle cell retinopathy, the ischemia develops from small arteriolar occlusions within the peripheral retina. This leads to increased levels of vascular endothelial growth factor (VEGF).
Our patient clearly has proliferative sickle-cell retinopathy O.S. She likely has regressed sickle-cell retinopathy in her right eye as well, as evidenced by the areas of retinal pigment hypertrophy.
Management of these patients is somewhat controversial. Panretinal photocoagulation (PRP) has been the mainstay therapy for many proliferative retinopathies, but there are no large-scale clinical trials in patients with sickle-cell retinopathy, which makes it difficult to provide a scientific basis for this form of treatment. Furthermore, up to 60% of sea fans spontaneously autoinfarct.1 So, close observation is usually recommended. For progressive disease and/or the presence of vitreous hemorrhage, PRP is still done.
Given that sea fan neovascularization results from increased levels of VEGF, Avastin, a VEGF inhibitor, may one day play an important role in the treatment of proliferative sickle cell retinopathy. When neovascularization is present, anti-VEGF drugs may allow for spontaneous regression to occur more easily, thus avoiding the need for more photodestructive procedures or more invasive surgery in eyes that develop vitreous hemorrhage and/or retinal detachment. For now, only one reported case shows the efficacy of Avastin for treating proliferative sickle-cell retinopathy.3
We elected to observe our patient with close follow-up. When she returned three weeks later, she reported intermittent episodes of the wispy floater O.S. The sea fan and fibrovascular changes O.S. were essentially unchanged. Because she had intermittent symptoms of vitreous hemorrhage, we administered an injection of Avastin O.S. and told her to return in one month.
Retina Quiz Answers: 1) c; 2) b; 3) c; 4) a.
1. Emerson GG, Harlan JB, Fekrat S, et al. Hemoglobinopathies. In: Ryan SJ, Schachat AP, eds. Retina, Vol. IIMedical Retina. 4th ed.
2. Sickle-cell anemia. TeensHealth. Available at: http://kidshealth.org/teen/diseases_conditions/blood/sickle_cell_anemia.html. (Accessed April 19, 2007)
3. Siqueira RC, Costa RA, Scott IU, et al. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand 2006 Dec;84(6):834-5.
