
In February 2007, I examined a 13-year-old Hispanic female, Jane. She was the youngest of Edwards three daughters, all of whom were my patients. At her previous examination, I noted that she had a corneal dystrophy, but it wasnt severe enough to merit anything more than monitoring. At this visit, Janes dystrophy still was not overly severe, but I was curious as to its origin. Biomicroscopic examination revealed cream-colored speckled deposits on the central anterior portion of each cornea.
History
I saw Janes sister Joanne two days later. Joanne had no corneal problems; however during that visit, Joanne told me about the corneal problems of both Edward and his other daughter, Donna. Edward also presented with the same corneal dystrophy. So, I referred Edward, Donna and Jane for anterior segment photography. Perhaps that could shed light on their similar presentations.
Diagnostic Data
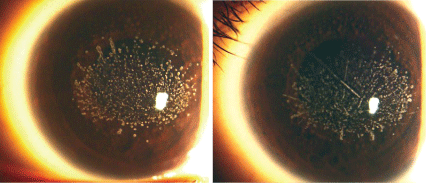
I compared the family members most recent records. Jane, whom Id just examined, presented with a manifest refraction of -1.50+3.00 x 90 O.D. and -1.50+2.50 x 90 O.S. (Snellen acuity: 20/40 O.D. and O.S.). She had deposits on her cornea (visible under a slit lamp) and received no treatment for them to date (figure 1).
1. Janes corneas at age 13, at her visit in 2007 (O.D. left, O.S. right). No treatment was initiated, but yearly follow-up was recommended. All photos courtesy: Harry Kachadorian
When I last saw Donna in 2004, her manifest refraction was -4.75D+3.50 x 70 O.D. and -1.25D+3.50 x 105 O.S. (Snellen acuity: 20/40- O.D., 20/30- O.S.) She also showed deposits on her cornea, but she had undergone phototherapeutic keratectomy (PTK) in 1998 (figure 2). However, her corneas still showed signs of deposition.
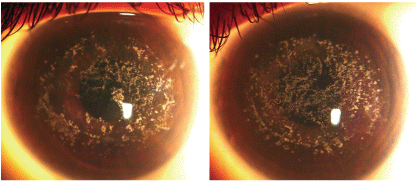
2. Donnas corneas at age 23 (O.D. left, O.S. right). Donna underwent PTK at age 14, in 1998. Since then, there has been a recurrence of granular deposits. This patient is being followed closely and presently uses ocular lubricants for recurrent corneal erosions when they occur. We are considering another treatment with PTK.
Joanne last presented for an exam in 2006. Her manifest refraction was -5.00D+0.75 x 095 O.D. and -4.50D+0.50 x 075 O.S. (Snellen acuity: 20/20 O.D. and O.S.) She was the only family member in my care not to present with deposits on the cornea.
Edward, the girls father, had last presented in 2003. At that time, his manifest refraction was -1.00D+1.50 x 133 O.D. and -1.75D +2.75 x 095 O.S. (Snellen acuity: 20/40+ O.D. and O.S.) His corneas also evidenced deposits, for which he had undergone PTK in 1998 (figure 3).
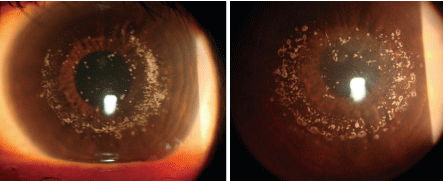
3. Edwards corneas at age 48 (O.D. left, O.S. right). He underwent PTK in 1998 as well. Unlike Donnas corneas, however, his remain fairly clear.
All four family members fundus examinations were unremarkable.
In each patient, anterior segment photos of both eyes revealed similar cream-colored deposits on the anterior central portion of the cornea. Edwards corneas demonstrated dramatically less deposits, due to the PTK hed undergone in 1998. Donna had also undergone the procedure at this time, though her corneas evinced more central deposits than her fathers.
Diagnosis
I diagnosed Jane, Donna and Edward with granular corneal dystrophy (GCD), a genetically inherited condition.
Treatment and Follow-Up
I referred Jane to the Rhode Island Hospital Eye Clinic recently for evaluation and blood testing. These samples were sent for testing to pin down the specific location of the genetic defect. Beyond that, we opted for yearly follow-up to monitor Janes progression.
Since Donna was exhibiting corneal deposits again and has had experienced symptoms of recurrent corneal erosion on several occasions, I recommended that she see our corneal specialist again to discuss the possibility of another treatment with PTK.
Also, Donna mentioned that her four-year-old daughter was already showing signs of granular dystrophy, so we scheduled an appointment for her to determine if she inherited the condition as well.
As for Edward, whose corneas were fairly clear, I recommended yearly follow-up examinations.
We hope to document this familys case of GCD by taking photos of Donnas four-year-old daughter, following up with Joanne (who evidenced no GCD) and photodocumenting Edwards brother, his sister and her children, who are also in the state.
Also, we are attempting to examine more family members. Edward has 15 siblingsand most of them live in
Discussion
Granular dystrophy is an anterior stromal dystrophy signaled by breadcrumb- or snowflake-like deposits that occur only in the central portion of the cornea. No deposits are seen in the periphery. They are composed of eosinophilic hyaline, but it is unknown which molecules the hyaline is derived from.1,2
The genetic defect occurs in chromosome 5q on the BIGH3 gene, which is a transforming, growth factor beta-induced gene (TGFBI).3-6 A transition of one base pair (e.g., cytosine to thymine, or guanine to adenine) would result in a change in an amino acid in the protein keratoepithelin. Mutations at different loci on the TGFBI gene may cause different dystrophies, such as granular corneal dystrophy, lattice dystrophy or Reis-Bucklers dystrophy.7 Edwin Stone, M.D., Ph.D., to whose lab Janes samples were sent, has found that granular, lattice and
Granular dystrophy is an autosomal-dominant inherited condition that usually displays 100% penetrance in offspring.1 It usually appears by the second decade of life, but vision may not be affected until the fourth or fifth decade.
Variants of this condition include juvenile granular dystrophy, in which numerous superficial opacities occur in the first decade of life and acuity is affected by the second decade of life, and Avellino dystrophy (named for the two Italian-American families with roots in Avellino, Italy, that manifested the dystrophy), which manifests both granular and lattice-like deposits, as well as a central haze.1
Granular dystrophy can occasionally cause recurrent corneal erosion (RCE), which can be managed like any other case of RCE with ocular lubricants, ointments, and if needed, bandage contact lenses. If the episodes of erosion still occur even with medical therapy, however, PTK may be effective in removing the superficial opacities. But, if a patient presents with deep lamellar or full thickness cornea deposits, corneal transplantation may be indicated.9
Edward and Donna both underwent PTK in 1998. Since then, Edwards GCD has been greatly minimized, but Donna has been experiencing bouts of RCE. Did the difference between their ages play a role in his successful outcome vs. her poorer outlook? In other words, does the patient need to be fully grown before PTK should be performed? Do males do better than females in general? The literature does explain that some patients demonstrate a recurrence of the dystrophy or require re-treatment.10 If the patient has significant visual disturbance because of the dystrophy, it may be beneficial to treat the patient, regardless of his or her age, and re-treat if needed later.
The prognosis is good for Edward, Jane and Donna as long as their vision is stable and their cornea epithelia are not eroding. Yearly examinations are essential in such cases, and it is important to monitor any relatives for signs of this condition.
Dr. Scharff is a clinical optometrist with Koch Eye Associates, a group multi-specialty ophthalmology and general optometry practice in
Dr. Scharff thanks Harry Kachadorian for his excellent photographs, Mark Heimmel, M.D., for arranging to have blood samples drawn for DNA analysis and for contacting a Guatemalan ophthalmologist to examine the extended family members, and Dr. Edwin Stone, M.D., Ph.D., for doing the DNA analysis.
1. Collin HB, Hendicott PL. Granular dystrophy of the cornea. Clin Exp Optom 1999 Sep-Oct;82(5):203-6.
2. Akhtar S, Meek KM, Ridgway AE, et al. Deposits and proteoglycan changes in primary and recurrent granular dystrophy of the cornea. Arch Ophthalmol 1999 Mar; 117(3): 310-21.
3. Kocak-Altintas AG, Kocak-Midillioglue I, Akarsu AN, Duman
4. Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mutat 2006 Jul;(27)7:612-25.
5. Cung le X, Ha NT, Chau HM, et al. Mutation analysis of the TGFBI gene in Vietnamese with granular and Avellino corneal dystrophy. Jpn J Ophthalmol 2004 Jan-Feb;48(1):12-6.
6. Zenteno JC, Santacruz-Valds C, Ramrez-Miranda A. Autosomal dominant granular dystrophy caused by TGFBI gene mutation in a Mexican family. Arch Soc Esp Oftalmol 2006 Jul;81(7):369-64.
7. Solari HP, Ventura MP,
8. Stone EM, Mathers WD, Rosenwasser GO, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Gen 1994 Jan; 6(1): 47-51.
9. Trattler W. Granular dystrophy. Available at: www.emedicine.com/oph/topic92 (Accessed April 6, 2007.)
10. Stewart OG, Pararajasegaram P, Cazabon J, Morrell AJ. Visual and symptomatic outcome of excimer phototherapeutic keratectomy (PTK) for corneal dystrophies. Eye 2002 Mar;16(2):126-31.
