 |
An eight-year-old Caucasian male presented with gradually progressive blurry vision in both eyes and the added difficulty seeing at night since early childhood. The patient reported that four of his maternal uncles experienced similar visual difficulties, which progressed with age. His ocular history is positive for a high myopic prescription, which began at age two and has progressed drastically every year. His medical history is positive for B-cell acute lymphoblastic leukemia in 2016, for which he received chemotherapy for two years. He is currently in medical remission and is closely monitored by his oncologist every three months. His social history is unremarkable.
On examination, best-corrected visual acuities were 20/60 OD, 20/80 OS with a prescription of -11.25 +3.00 x 107 OD, -9.25 +2.75 x 0975 OS. The patient’s extraocular motility was full and smooth. Confrontation visual fields revealed a 360˚ peripheral constriction in both eyes. Pupils were equal, round and slowly reactive to light with no afferent pupillary defect in either eye.
Color vision, measured with Ishihara plates, was reduced for both eyes (7/10 OD, 8/10 OS). Intraocular pressures (IOPs) were 20mm Hg OD and 21mm Hg OS. Anterior segment health was unremarkable for both eyes. A dilated fundus examination revealed changes. Optical coherence tomography (OCT) and visual fields were also performed.
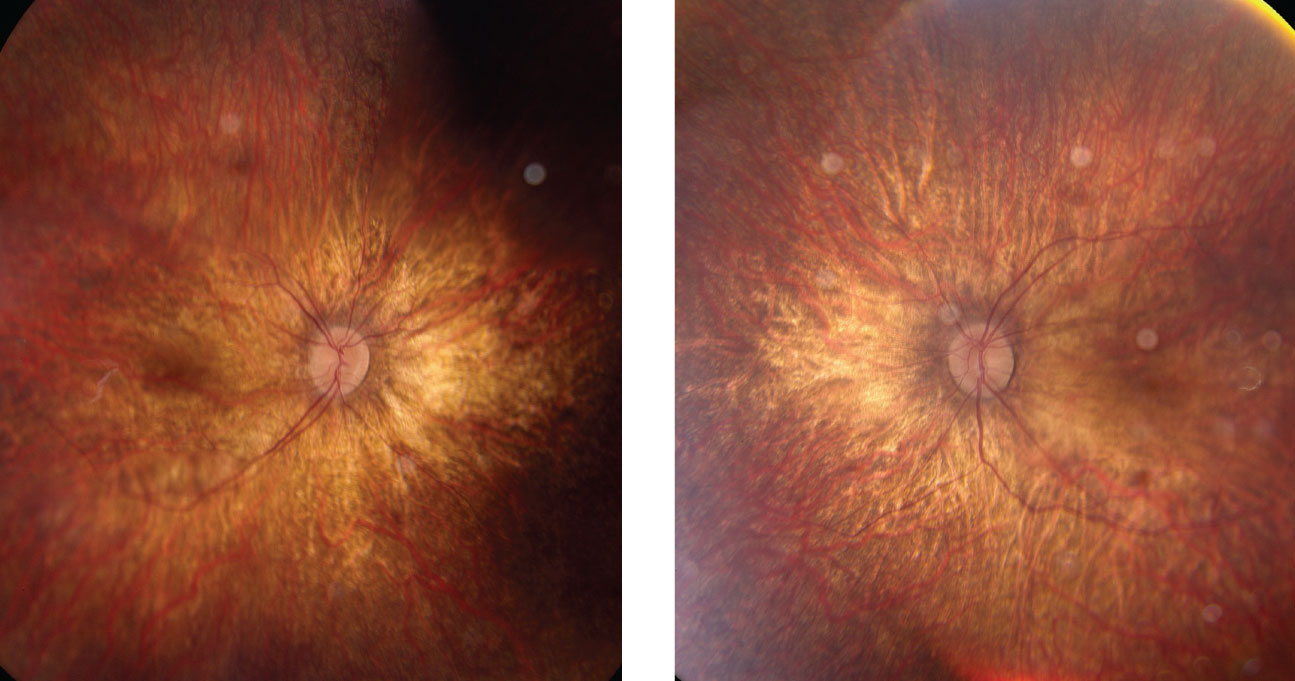 |
| Right and left eyes of our young patient showing an ultra-widefield view of the posterior pole and periphery. What pathology could explain his blurry vision? Click to enlarge. |
Take the Retina Quiz
1. Which statement best describes the fundus appearance of our patient?
a. Blonde fundus with a loss of the retinal pigment epithelium and choroid.
b. Vascular attenuation with disc pallor and increased choroidal show.
c. Diffuse outer retinal atrophy with exposed sclera, sparing the macula.
d. Vascular attenuation with surrounding angioid streaks.
2. What is the likely diagnosis for this patient?
a. Fundus albipunctatus.
b. Retinitis pigmentosa.
c. Choroideremia.
d. Gyrate atrophy.
3. What is the underlying etiology of this condition?
a. Infectious.
b. Congenital.
c. Inflammatory.
d. Hereditary.
4. What is the most common first symptom of this condition?
a. Progressive blurry vision.
b. Nyctalopia.
c. Reduced color vision.
d. Loss of peripheral vision.
5. Based on the presentation, what is the appropriate next step?
a. Monitor annually.
b. Refer for an electroretinograph and genetic testing.
c. Treatment with vitamin E supplements.
d. Refer the patient to a low vision specialist.
For answers, see below.
Diagnosis
Based on the history and clinical presentation, the patient was tentatively diagnosed with retinitis pigmentosa (RP) and sent for a full-field electroretinogram (ERG) as well as genetic testing to confirm the diagnosis. We also considered the possibility of choroideremia based on the fundus appearance; however, the ERG results revealed diffuse flattening of both the A and B waveforms, indicating reduced functioning of the photoreceptors and inner retinal cells (i.e., bipolar and Mueller cells) more consistent with RP.1 In addition, genetic testing came back positive for a mutation in the RP2 gene. Based on the ERG findings and genetic testing, along with a strong family history of RP, we confirmed that our patient has x-linked recessive retinitis pigmentosa.
Retinitis pigmentosa is a group of genetic disorders with varying modes of inheritance, from autosomal dominant to x-linked recessive. It is a familial condition that causes bilateral, progressive vision loss typically in individuals between nine and 19 years old with no sex or race predilection.2 Symptoms of RP include night blindness (most common), loss of peripheral vision, reduced color vision and blurred vision.3,4 Clinical signs of RP include a triad of a waxy optic disc pallor, vessel attenuation and perivascular bone-spicule pigmentation in the mid-periphery.3 Additional signs include diffuse RPE atrophy, cystoid macular edema, optic disc drusen (10% of cases), epiretinal membrane, vitreous condensation, keratoconus and posterior subcapsular cataracts (35% to 51% of cases).3 To date, this patient has presented with optic disc pallor and vessel attenuation with diffuse thinning of the retina secondary to myopic degeneration.
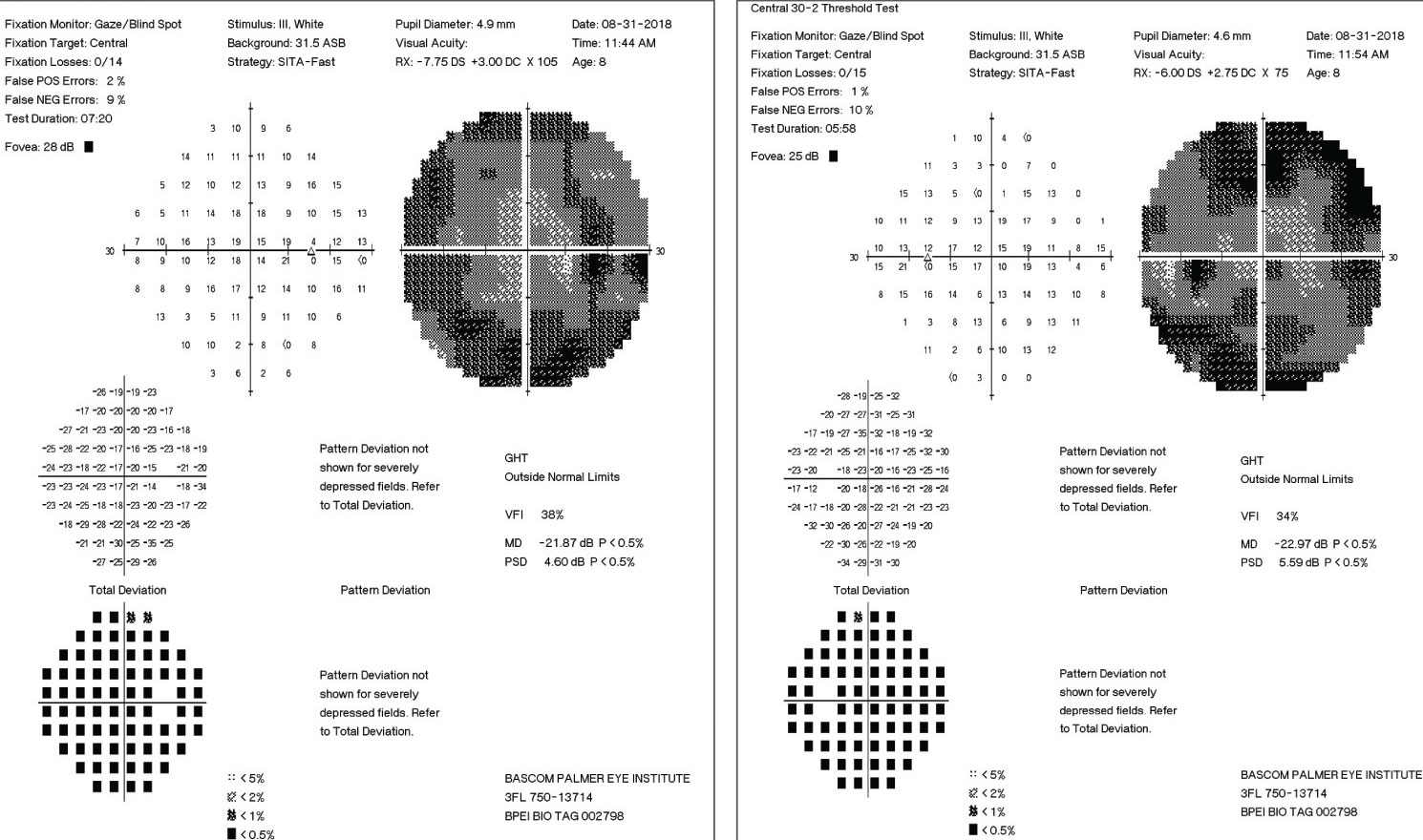 |
| These 30-2 visual fields showing significant loss of the peripheral fields in each eye. Click to enlarge. |
Discussion
An estimated 100,000 Americans have RP as a result of several genetic mutations.5 More than 60 mutated genes may cause RP, 20 of which are associated with the autosomal dominant form of this disorder.5 In contrast, mutations in about six genes contribute to the x-linked form of this condition. In fact, mutations in the RPGR and RP2 genes account for a majority of X-linked RP individuals, as is the case with this patient.5 The RP2 gene product mimics the human cofactor C-protein involved in beta-tubulin folding.3,5 Mutations in this gene result in an accumulation of incorrectly folded tubulin isoforms resulting in subsequent cellular death and progressive retinal degeneration.3,5
Imaging
Multimodal imaging can help diagnosis. In RP patients, fundus autofluorescence typically displays an abnormal hyperautofluorescence as a result of lipofuscin accumulation from increased phagocytosis of photoreceptor outer segments.2 The presence of a hyperautofluorescent macular ring is classic in RP patients and represents the transitional zone between healthy and dying photoreceptors.2,3 Over time, this ring constricts with the advancement of this disorder and can be used as an indicator of progression. OCT can also help to identify signs of RP. With the progression of this disorder, OCT shows a disruption of the ellipsoid band and a thinning of the outer nuclear layer due to photoreceptor cell loss.2,6 This, in conjunction with full-field ERG as well as genetic testing, contributes pieces to the puzzle and allows for a more comprehensive diagnosis.
For a majority of cases, RP is an isolated disorder (simple or non-syndromic). However, it can be associated with systemic conditions, such as Usher’s syndrome, Bardet-Biedl syndrome, Cockayne’s syndrome, Alstrom’s disease abetalipoproteinemia and more.6 In this case, the patient only had an isolated form of this disorder with no evidence of systemic associations.
Treatment
RP presently has no cure. In 2012, three peer-reviewed clinical studies reported that a combined regimen of 15,000 IU of vitamin A palmitate, oily fish and lutein can help slow the rate of visual acuity lost per year by RP patients by preserving retinal function.2,3 Oily fish contributes towards a rich omega-3 diet, of which docosahexaenoic acid (DHA) is a major constituent. This treatment method, however, is controversial in children and no specific studies focus on the more-aggressive x-linked RP.3 Nevertheless, a clinical trial evaluating the benefits of DHA in people with x-linked RP is currently underway.6
For advanced cases of RP, the Argus II retinal prosthesis, a surgically implanted device, is being used to deliver electrical stimulation to the retina to restore visual perception.2,3 However, the effectiveness of this device is yet to be demonstrated. Retinal stem cell treatment for RP patients is in its Phase I/IIa safety trial.6 The basis of this treatment is to inject retinal progenitors (similar to stem cells) into the vitreous with the hopes of rescuing and reactivating the recipient’s photoreceptors before they die.6 However, this study is still in its nascency and has yet to be tested for all forms of RP.
Our patient was referred to the Miami Lighthouse for the Blind for solutions to optimize visual performance and is being monitored on an annual basis with visual fields and OCT imaging.
Dr. Jayasimha is doing a residency in ocular disease at Bascom Palmer Eye Institute in Miami.
1. Creel D. The electroretinogram and electro-oculogram: clinical applications. Webvision: The Organization of the Retina and Visual System. webvision.med.utah.edu/book/electrophysiology/the-electroretinogram-clinical-applications. July 14 2015. Accessed. September 9, 2018. 2. Fahim A, Daiger S, Weleber R. Retinitis pigmentosa overview. Gene Reviews. www.ncbi.nlm.nih.gov/books/NBK1417/. March 21, 2013. Accessed September 9, 2018. 3. Hamel C. Retinitis pigmentosa. OJRD. ojrd.biomedcentral.com/articles/10.1186/1750-1172-1-40. October 11 2006. Accessed Sept. 9, 2018. 4. Iannaccone A, Berdia J. Retinitis pigmentosa. National Organization of Rare Disorders. rarediseases.org/rare-diseases/retinitis-pigmentosa. July 05, 2016. Accessed Sept. 9, 2018. 5. Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011 Jun;12(4):238-49. 6. Klassen H. Groundbreaking stem cell treatment for retinitis pigmentosa. JCyte. jcyte.com/our-mission. July 01, 2018. Accessed September 9, 2018. |
Retina Quiz Answers:
1) b; 2) b; 3) d; 4) b; 5) b.
