 History
History
A 53-year-old white male presented complaining of poor night vision, which had been progressively worsening over a six-month period. His systemic history was remarkable for hypertension. He had no known allergies.
Diagnostic Data
The patients best-corrected visual acuity was 20/25 O.U. at distance and near. Extraocular muscles, external testing and pupils were normal.
His confrontational visual fields were slightly more constricted in the right eye. His refractive changes were negligible.
His anterior segments were normal by slit lamp exam, and his intraocular pressures measured 14mm Hg O.U. The significant posterior segment findings are illustrated in the photographs.
Additional tests included electroretinogram (ERG), electrooculogram (EOG), dark adaptometry and photodocumentation.
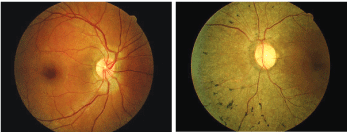
A 53-year-old male who complained of poor night vision (O.D. left, O.S. right).
Your Diagnosis
How would you approach this case? Does this case require any additional tests? What is your diagnosis? How would you manage this patient? Whats the likely prognosis?
Dr. Gurwood acknowledges and thanks Robert Dusak, O.D., for contributing this case.
The Diagnosis
The diagnosis in this issue is a rare case of unilateral retinitis pigmentosa (RP). RP is a clinically and genetically heterogeneous group of inherited retinal disorders characterized by progressive photoreceptor and retinal pigment epithelium (RPE) degeneration.1-15 Visual impairment usually manifests as night blindness, visual field loss, and in some cases, central visual dysfunction.
The age of onset for associated visual impairment ranges from infancy to adulthood.1-15 Visual compromise caused by RP may be as mild as unnoticed sectorial visual field loss or as severe as profound peripheral visual field loss (tunnel vision), with varying degrees of central macular depreciation over time.2
Retinitis pigmentosa is the most common form of retinal degeneration; it affects approximately 1.5 million people worldwide.7,11 The overall incidence in the
About half of the affected individuals in the
Environmental (non-genetic) causes of RP are difficult to determine. After extensive history and evaluation, many patients are found to have other relatives who had RP, night blindness or blindness of unknown etiology.
The term "retinitis pigmentosa" is a misnomer. The retinal inflammation implied by the nomenclature is not a prominent feature of the pathophysiology.14 Today, RP includes a wide spectrum of disorders with diverse chromosomal, metabolic and morphologic findings. Most associated disorders are genetically determined and cause progressive degeneration of the photoreceptors, which in turn yields a combination of visual field defects, altered night vision and compromised central vision.2
ERG is the most common electrophysiologic method of diagnosing RP.2 In this procedure, photoreceptor cells are either dark-adapted (scotopic ERG) or adapted to a specific level of light (photopic ERG), and then are stimulated with a brief flash of light. The aggregate electrical response of the retina is recorded extraocularly with a contact lens electrode placed on the cornea.2 The responses are amplified and displayed on an oscilloscope.2
The scotopic ERG selectively measures the response of the rod photoreceptor cells, while the photopic ERG measures the response of the cones. In typical RP, the rod/cone disease initially presents as alterations of the scotopic ERG and shows a proportional loss of the photoreceptor cell and post-photoreceptor components. Patients with the early stages of retinitis pigmentosa have ERGs that are reduced in amplitude and delayed temporally.2
An EOG is an indirect measure of the standing potential of the eye.16 In general, the electrooculogram is abnormal in diffuse hereditary rod/cone degenerations of the retina.16,17 The EOG, in most cases of RP, is abnormal when the ERG is abnormal.17 In fact, in almost all cases, the EOG findings correlate with the ERG findings.16 In most of the degenerative and dystrophic disorders, the EOG serves as an adjunct test for the ERG. In a clinical situation, an EOG is unnecessary if the ERG is found to be diagnostic of RP.17
Pattern reversal visual evoked response (PVER) has demonstrated to be useful as an objective evaluation of visual function in RP patients whose central vision is preserved.17
There is no effective treatment for retinitis pigmentosa, despite longstanding interest in slowing or arresting the process.17 Genetic and psychological counseling can be helpful, and low vision rehabilitation services offer promise. However, vitamin therapy, gene therapy, cell transplantation, neuroprostheses and new pharmacotherapies remain under investigation.
1. Bressant DA, Payne AM, Snow BE, et al. Importance of the autosomal recessive retinitis pigmentosa locus on 1q31-q32.1 (RP12) and mutation analysis of the candidate gene RGS16 (RGS-r). J Med Genet 2000 May;37(5):384-7.
2. Weleber R. Retinitis pigmentosa and allied disorders. In: Ryan S, Ogden T, Schachat A, eds. Retina, 2nd ed.
3. Berson E. Retinitis Pigmentosa and Allied Diseases. In: Albert DM, Jakobied FA, eds. Principles and practice of ophthalmology: clinical practice, vol 3, 2nd ed.
4. Strettoi E, Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci U S A 2000 Sep 26;97(20):11020-5.
5. Wong F. Investigating retinitis pigmentosa: a laboratory scientists perspective. Prog Ret Eye Res 1997 July;16(3):353-73.
6. Scholl HP, Kremers J. Large phase differences between L-Cone- and M-Cone-driven electroetinograms in retinitis pigmentosa. Invest Ophthalmol Vis Sci 2000 Sep;41(10):3225-33.
7. Applebury M., Hargrave PA. Molecular biology of the visual pigments. Vision Res 1986;26(12):1881-95.
8. Boughman JA,
9. Fishman GA. Retinitis Pigmentosa. Genetic percentages. Arch Ophthalmol 1978 May;96(5):822-6.
10. Berson EL. Retinitis pigmentosa. The Friedenwald lecture. Invest Ophthalmol Vis Sci 1993 Apr;34(5):1659-76.
11. Berson EL. Nutrition and retinal degenerations. Int Ophthalmol Clin 2000 Fall;40(4):93-111. Review.
12. Baumgartner WA. Etiology, pathogenesis, and experimental treatment of retinitis pigmentosa. Med Hypotheses 2000 May;54(5):814-24.
13. Newsome DA. Retinitis Pigmentosa, Ushers Syndrome, and Other Pigmentary Retinopathies. In: Newsome DA, ed. Retinal Dystrophies and Degenerations.
14. Carr RE. The Generalized Heredoretinal Disorders. In: Regillo CD, Brown GD, Flynn HW Jr., eds. Vitreoretinal Disease: The Essentials.
15. Slamovits TL, Heckenlively JR, Abrams GW, et al. Clinical applications of visual electrophysiology and psychophysics. Retina and Vitreous.
16. Arden GB, Fojas MR. Electrophysiological abnormalities in pigmentary degenerations of the retina. Assesment of value and basis. Arch Ophthalmol 1962 Sep;68:369-89.
17. Paranhos FR, Katsumi O, Arai M, et al. Pattern reversal visual evoked response in retinitis pigmentosa. Doc Ophthalmol 1998-1999;96(4):321-31.
