 |
History
A 56-year-old man presented with a chief complaint of blurred vision for the previous six months. He explained that his spectacles were old and that he wanted new ones with an updated prescription.
He had no known previous ocular or systemic history and reported no allergies.
Diagnostic Data
His best corrected visual acuities were 20/30 OD and 20/30 OS at distance and 20/40 OU at near. External examination was normal and we saw no evidence of afferent pupillary defect.
Refraction revealed symmetrical, spherical myopia with a +0.50D change in the add yielding 20/25 acuity at near. No improvement could be achieved at distance.
The anterior segments of both eyes were in good health and applanation intraocular pressures measured 19mm Hg OU. The pertinent dilated fundus finding OU is demonstrated in the photographs.
Your Diagnosis
Does this case require additional tests? How would you manage this patient? What is the likely prognosis?
Discussion
Additional tests/procedures included laser interferometry which demonstrated maximal visual acuity of 20/30, cover testing which revealed five prism diopters of exophoria at distance and 10 prism diopters of exophoria at distance, stereo testing which demonstrated 50 seconds of contour stereo at 16 inches with the presence of global stereo and visuoscopy which demonstrated steady bifoveal central fixation. The optic discs and posterior poles were photodocumed.
The diagnosis in this months issue is ocular albinoidism. Albinism is a genetic condition associated with defects in melanin production causing underdevelopment of the visual system and fovea, reduced retinal cell numbers and abnormal routing of ganglion cell nerve fibers within the optic chiasm and visual pathway.1 The prevalence of all forms of albinism varies with estimates at approximately 1/17,000 persons.2 This suggests that one in 70 people carry a gene for albinism in one form or another.2
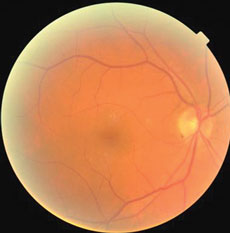 | 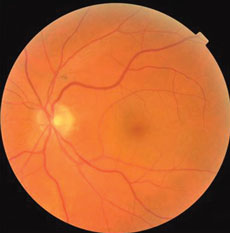 |
| These fundus photos show the eyes of a 56-year-old male patient who has suffered from blurred vision for six months. Can the images help you identify the cause of his condition? | |
Oculocutaneous albinism (OCA) is a group of inherited disorders of melanin biosynthesis characterized by reduction in pigmentation of the hair, skin and eyes.2 The clinical spectrum of OCA has a range with OCA1A being the most severe type demonstrating a complete lack of melanin production throughout life.2 The milder forms include OCA1B, OCA2, OCA3 and OCA4 with each showing some pigment accumulation over time.2 The clinical manifestations of OCA include degrees of congenital nystagmus, iris hypopigmentation and translucency, varying degrees of strabismus, reduced stereoscopic vision, reduced pigmentation of the retinal pigment epithelium, foveal hypoplasia, reduced visual acuity usually (20/60 to 20/400), varying refractive errors, color vision impairment and prominent photophobia.2
Oculocutaneous albinism has systemic syndromic connections.3-5 Hermansky-Pudlak syndrome (HPS) is an autosomal recessive disorder characterized by oculocutaneous albinism, platelet dysfunction and multisystem tissue lysosomal ceroid deposition.3,4 It is commonly seen in the Puerto Rican population.3 Chediak-Higashi syndrome is a rare autosomal recessive disorder where patients show hypopigmentation, recurrent infections, mild coagulation defects and varying neurologic problems.5
Ocular albinism (OA) is characterized as a group of genetic disorders presenting with reduced pigmentation of the eye in association with variable degrees of decreased visual acuity, nystagmus, strabismus and photophobia, in the setting where pigmentation of skin and hair is relatively normal.6
Incomplete albinism or albinoidism is characterized by variable manifestations of the condition owing to a genetic variation.7
All four types of OCA have been established as inherited autosomal recessive disorders.2 Studies have shown that ocular albinism may constitute a clinically mild presentation of oculocutaneous albinism due to mutations in either the TYR (OCA1) or OCA2 (P) genes.6 The genetic mutation responsible for HPS remains unknown.4 The CHS1/LYST gene has been associated with Chediak-Higashi syndrome.5 Mutations in the OA1 gene on the short arm of the X chromosome are known to cause X-linked ocular albinism known as the Nettleship-Falls variant.7,8
The common denominator of decreased vision in patients with albinism is decreased foveal volume and macular hypoplasia.1,9-11 This finding is compatible with a reduction or loss of ganglion cell numbers.1,2 New research suggests that an observation of no foveal pit be known by the specific term as fovea plana.9 Patients may be “fovea plana” without the effects of foveal hypoplasia.9,10 As the foveal region contains the highest cell density in the human retina, a large area of the visual cortex is dedicated to its representation.10,11 In cases of both aniridia and albinism, the fovea does not properly develop causing its corresponding cortical representations to be reduced.1,11 Analysis reveals that regionally specific decreases in grey matter at the occipital poles in patients with albinism corresponds to the cortical representation of the central visual field and correlates to most of the compromises in visual function.1
In patients with albinism there are additional chiasmal irregularities and alterations in the hemispheric projections.1,11 Misrouting of the fibers at the optic chiasm, where the majority of fibers cross to the contralateral side, produces an abnormal decussating pattern.9-11 This reflects the disturbance within the retina controlled in part by the by the dysfunction of melanin metabolism.12 It also is the predominant reason many of these patients lack true binocularity and often present with some strabismus.
Foveal hypoplasia is related to poor fundus pigmentation in the region.13 Melanocytes are pigment-producing cells residing primarily in hair follicles, the skin and eye.13 Pigmentation is achieved inside melanocytes when melanin is produced and sequestered within specialized organelles known as melanosomes.13 The enzyme tyrosinase, a type I membrane glycoprotein, plays a key role in the initiation of the melanogenesis process in humans.14-16 Mutations in the human tyrosinase gene cause the tyrosinase negative form of oculocutaneous albinism.15
Whenever a patient with fair skin and minimal funduscopic pigmentation is observed, with or without the presence of compromised visual acuity, the possibility of albinism should be considered.1-11 A complete family history including investigation of family photographs may help uncover a genetic link previously not realized. Since albinism has linkage to systemic syndromes, its existence cannot be dismissed simply because the patient reports no clinical symptoms. In cases of high suspicion, referral for genetic counseling/testing (tyrosinase hair bulb assay) may be indicated, for both the welfare of the patient and to understand the risk to potential future generations.2,16
Ocular management for all forms of albinism begins with appropriate spectacle correction. Here, the triple consideration includes refractive error, photophobia and the harmful rays of the sun. Spectacle corrections should be designed considering either sun specific lenses, photochromic lenses such as Transistions (Transitions Optical Inc.), Photogray (Corning Optical/Winchester optical) or the option for protective UV shields (CP films) or NOIR shields (NOIR medical technologies). Prescribing the maximum correction tolerable, especially in younger patients will maximize visual acuity and minimize the amblyogenic effects of large, astigmatic or asymmetric corrections. In cases of strabismus (with or without an accommodative or refractive component) or where visual acuity cannot be corrected to the values obtained via laser interferometry (maximum projected foveal potential), strabismic amblyopia must be considered, with an appropriate surgical referral or referral for binocular evaluation and training.
In cases where traditional spectacles do not alleviate visual disabilities, low vision rehabilitation along with orientation and mobility rehabilitation services exist to provide magnifying devices (full field full diameter microscopes, prism half eye microscopes, stand magnifiers, hand-held microscopes, telemicroscopes, telescopes and closed circuit televisions) as well as supportive aides (acetate filters to increase the contrast of printed materials, large print books, enlarged clocks etc…). The practice of low vision is an art form, requiring a combination of knowledge and imagination. It is best accomplished using a team approach between a trained prescribing practitioner and a rehabilitation specialist.
The nystagmus sometimes seen in patients with albinism is often termed “searching.”17,18 Research supports that acuity development in these cases is directly related to the visual sensory defects and not the nystagmus.17,18 The nystagmus develops secondary to the poor ability to stabilize fixation as a result of the compromised fovea. The characteristics of searching nystagmus include bilaterality, conjugate horizontal uniplanar movements and an accelerating slow phase.
Patients with any form of albinism must have regular skin evaluations and be informed of the need for strict and consistent sun protection strategies.1,20,21
The traditional treatment for Chediak-Higashi syndrome is a bone marrow transplant to remedy the hematologic and immune defects.5 Unfortunately, nothing can be done to alter the neurologic problems.5 The treatment for Hermansky Pudlak syndrome ranges from transfusion of platelets to medicating with antifibrinolytics to clotting factor replacement therapy and stem cell transplantation.3,22
|
