A 40-year-old white female presented with a chief complaint of tearing and some visual blur in her right eye that persisted for a few weeks. She also reported occasional diplopia.
The patient had been diagnosed with Wegener’s granulomatosis (WG) a year earlier. Otherwise, her medical and family histories were unremarkable.
Her current medications included 20mg oral prednisone b.i.d. and a monthly infusion of 1,000mg/meter2 of Cytoxan (cyclophosphamide, Bristol-Myers Squibb).
Diagnostic Data
Her entering visual acuity measured 20/25 O.D. and 20/20 O.S. External examination revealed no abnormalities. Her pupils were equally round and reactive to light and accommodation, with no afferent defect O.U. Extraocular muscles were full, accurate, smooth and extensive in both eyes.Additionally, her confrontation fields were full O.U.
Refraction yielded no improvement in visual acuity. Biomicroscopy showed no abnormalities in the anterior segment O.U. Intraocular pressure measured 24mm Hg O.U.
The right lens showed the early formation of a posterior subcapsular cataract. Dilated fundus examination revealed a healthy optic nerve head with 0.3 x 0.3 cups in addition to well-perfused rim tissue, a healthy nerve fiber layer and macula, and unremarkable peripheral structures.
Treatment and Follow-up
Given the findings, we prescribed Travatan Z (travoprost, Alcon) at bedtime to reduce her intraocular pressure. Additionally, we scheduled her for a three-week follow-up.
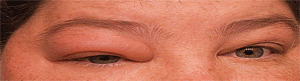
1. Our patient exhibited orbital inflammation and proptosis that was seen primarily in the right eye.
At the follow-up visit, our patient noted an increase in all previously reported symptoms. And, although the eye looked normal, her intraocular pressure had increased to 30mm Hg O.D. and 18mm Hg O.S. We decided to add Combigan (brimonidine and timolol, Allergan) b.i.d. to her regimen. We then scheduled the patient for another three-week follow-up.
Just two weeks later, the patient called our office, complaining of increased discomfort. We asked her to come in before her scheduled follow-up appointment. When she arrived at the office, she exhibited a noticeable proptosis (figure 1) with restricted motility in the right eye. Additionally, her intraocular pressure now measured 48mm Hg O.D.
We ordered an orbital magnetic resonance imaging scan (figure 2), which revealed bilateral orbital granulomas (O.D. > O.S.). After consulting the patient’s rheumatologist, we increased her dosage of oral prednisone to 60mg per day and recommended a 33% increase in Cytoxan. Also, we added Azopt (brinzolamide, Alcon) b.i.d. O.D. We again scheduled her for a three-week follow-up.
At this follow-up visit, the patient reported an improvement in both pain and discomfort; however, the increased oral steroid dosing resulted in weight gain, insomnia, poorly controlled hypertension and anxiety.
To avoid the excess steroid use, we selectively injected 1mL triamcinolone acetate (40mg/mL) into the lateral aspect of her right orbit.
In just one week, the orbital granulomas regressed (figure 3). Additionally, her intraocular pressure dropped to 16mm Hg O.U. Afterward, the patient remained asymptomatic on a reduced oral steroid regimen. Also, her intraocular pressure remained the same with the use of only topical medications.
Discussion
WG is an arteriolar vasculitis that was first documented in 1931.1 There are approximately 24 to 157 cases of WG per one million individuals in the United States, with three to 14 new cases per one million individuals reported each year.1
Typically, WG presents with multiple organ involvement. Its etiology is primarily unknown, although there have been reports of a genetic predisposition.2 While the disease is seen in both children and adults, the mean age of detection is 41 years.3
The classical form of WG exhibits a triad of necrotizing inflammation in the upper respiratory tract, glomerulonephritis and systemic vasculitis. In one form of WG, systemic involvement is limited to the respiratory tract with an absence of renal involvement.
The pathogenesis is not clearly understood, but autoimmunity is a widely accepted cause. A review of other vasculitides may be helpful in understanding the pathogenesis of WG. Churg-Strauss syndrome and polyarteritis nodosa (PAN) are two such conditions that present similarly to WG, both affecting small to medium arterioles.1,2
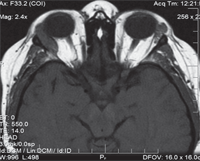
2. Orbital magnetic resonance imaging scan revealed a pronounced granuloma in the right orbit. In addition, we noted the presence of a smaller granuloma in the left lateral orbit.
Immunofluorescence studies show the presence of antineutrophil cytoplasmic auto antibodies (ANCA) in a majority of patients with such cases of vasculitis.4 There seem to be two different antigens involved—proteinase 3 (PR3) and myeloperoxidase. These antigens are primarily located in the cytoplasm in patients with WG (referred to as cANCA) and in the nuclear and perinuclear areas in patients with PAN.5,6
Patients with WG have an increased number of neutrophils that express constitutive PR3. ANCA can activate these neutrophils to release free radicals and lytic enzymes, which can damage vascular endothelial cells and lead to necrosis.
Recent studies in a mice model also have shown the need for PR3 for vasculitis to take place.5 Although measurement of anti-PR3cANCA gives a more practical way to diagnose and monitor the disease, the specificity of ANCA for WG is a concern.
Studies show sensitivities ranging from 75% to 90%, but anti-PR3cANCA has become an adjunct, if not primary, method of monitoring the disease. Still, biopsy of the granuloma remains the most reliable diagnostic method.6
Incidence of ophthalmic involvement can occur in both forms of WG and presents in 28% to 60% of the cases.7,8 In many cases, ocular involvement actually has lead to the identification of previously undiagnosed WG.
A survey of 140 confirmed cases showed that the most common ophthalmic manifestations were orbital inflammatory disease and necrotizing sclerokeratitis.8 Also, a review of nine cases from India showed that necrotizing scleritis and peripheral keratopathy were the most common ophthalmic presentations of WG.9
Other ophthalmic manifestations reported include scleritis, episcleritis, conjunctivitis, corneal ulceration, uveitis, retinal vasculitis, retinal vascular occlusions, retinal detachments, optic neuropathy, cellulitis, and obstruction of the nasolacrimal duct.2,10,11 Granulomatosis can occur within the orbit or can infiltrate from the nasal sinuses, resulting in proptosis and secondary ophthalmic complications.2
In one case report, orbital involvement resulted in paralysis of the third, fourth and sixth cranial nerves as well as the first division of the fifth cranial nerve and ischemic compression of the optic nerve.3 Ischemic conditions resulting from vasculitis of the scleral blood vessels can lead to necrosis and even perforation.
In a retrospective study of 49 WG patients in the United Kingdom, 28 individuals exhibited ocular involvement.12 Of these, 21 had focal involvement with conjunctivitis, episcleritis, scleritis, keratitis, iritis or retinitis.12 An additional seven patients had orbital involvement.12
Further, of the 28 patients with ocular involvement, three died from the disease. This study also indicated that early diagnosis and treatment can result in a better visual prognosis.
In another case from the UK, the authors reported perhaps the first case of extraocular muscle myositis associated with WG as the initial presenting sign.13 Interestingly, a survey of Slovenian patients examined from 2003 through 2008 indicated that associated ocular manifestations served as the primary diagnosis in 46.7% of WG cases.
Treatment of the underlying systemic condition also helps control the ocular manifestations that do not respond to topical agents. Surgical decompression may be of value in serious orbital involvement with optic neuropathy. Despite the use of systemic immunosuppressants, irreversible ischemic neuropathy has been reported.3 So, eye care providers must be vigilant and aggressive during treatment to avoid permanent vision loss.
Management of WG is accomplished in conjunction with rheumatologists, pulmonologists, internists and oncologists. A multispecialty approach is critical, because the effects of the disease are so widespread. The mainstay therapy involves immunosuppression with cyclophosphamide and steroids. Cyclophosphamide—a chemotherapeutic agent—and prednisone have been shown to be effective in controlling the disease and its associated ocular manifestations.14
In a case like ours, where the patient already is on an oral steroid, the ocular side effects of the systemic condition must be included in the differential diagnosis before implicating the steroid.
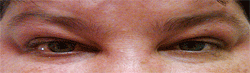
3. Our patient showed marked improvement following an adjustment of her medication and a triamcinolone acetate injection.
Although the patient initially appeared to be a steroid responder, the ocular hypertension was, in fact, largely caused by the orbital granuloma impeding trabecular outflow. Orbital inflammation was caused by the primary granulomas or a spread of the granulomas from the nasal sinuses.3
Either way, proptosis and restricted motility are common clinical signs of WG. Optic neuropathy may accompany these symptoms, but was not documented in our patient. Although paralysis of the extraocular muscles has been reported in similar cases, we believe the restrictive ocular motility in our patient probably is a mass effect rather than a frank paralysis.
Steroid use is an essential element of WG management, but the risk of steroid-induced glaucoma is a distinct possibility. So, the use of ocular hypotensive medications may be helpful in controlling this complication, as illustrated in this case.
The chronic nature of WG necessitates long-term steroid use, which can cause multiple side effects that could be life threatening. Therefore, WG patients with a long-standing history of oral steroid use often require constant monitoring and intervention. Localized treatments with triamcinolone acetate may be added to control the orbital granuloma, relieve patient symptoms, and reduce the need for excess oral steroid use.
WG is a serious systemic disease with multiple organ involvement, including the eyes. Patients should be closely monitored for any ocular involvement and treated aggressively to avoid ocular morbidity. Eye care providers should be vigilant, because ocular manifestations sometimes may be the primary and/or only sign of WG.
Although ocular manifestations may take various forms, WG should be included in the differential diagnosis—especially when there is orbital involvement or scleral necrosis. Localized treatment with triamcinolone acetate may be added to the systemic treatment to control ocular symptoms and avoid excess oral steroid use, particularly when orbital granuloma is involved.
Dr. Ananthan-Nair is in private practice in Debary, Fla. Dr. Barber is in private pratice in Orange City, Fla. They have no direct financial interest in any of the products mentioned.
1. Mahr AD, Neogi T, Merkel PA. Epidemiology of Wegener’s granulomatosis: Lessons from descriptive studies and analyses of genetic and environmental risk determinants. Clin Exp Rheumatol. 2006 Mar-Apr;24(2 Suppl 41):S82-91.
2. Harman LE, Margo CE. Wegener’s granulomatosis. Surv Ophthalmol. 1998 Mar-Apr;42(5):458-80.
3. Chua J, Lim L. Systemic Wegener’s granulomatosis with severe orbito-ocular involvement. Singapore Med J. 2008 Oct;49(10):e259-62.
4. van der Woude FJ, Rasmussen N, Lobatto S, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985 Feb 23;1(8426):425-9.
5. Savage CO, Harper L, Holland M. New findings in pathogenesis of antineutrophil cytoplasm antibody-associated vasculitis. Curr Opin Rheumatol. 2002 Jan;14(1):15-22.
6. Mansi IA, Opran A, Rosner F. ANCA-associated small-vessel vasculitis. Am Fam Physician. 2002 Apr 15;65(8):1615-20.
7. Pakrou N, Selva D, Leibovitch I. Wegener’s granulomatosis: ophthalmic manifestations and management. Semin Arthritis Rheum. 2006 Apr;35(5):284-92.
8. Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegener’s granulomatosis. Ophthalmology. 1983 Mar;90(3):279-90.
9. Biswas J, Babu K, Gopal L, et al. Ocular manifestations of Wegener’s granulomatosis. Analysis of nine cases. Indian J Ophthalmol. 2003 Sep;51(3):217-23.
10. Pahor D, Gracner B, Gracner T, Pahor A. Ocular symptoms as the initial signs of Wegener’s granulomatosis. Klin Monbl Augenheilkd. 2009 May;226(5):409-13.
11. Spalton DJ, Graham EM, Page NG, Sanders MD. Ocular changes in limited forms of Wegener’s granulomatosis. Br J Ophthalmol. 1981 Aug;65(8):553-63.
12. Sadiq SA, Jennings CR, Jones NS, Downes RN. Wegener’s granulomatosis: The ocular manifestations revisited. Orbit. 2000 Dec;19(4):253-61.
13. Salam A, Meligonis G, Malhotra R. Superior oblique myositis as an early feature of orbital Wegener’s granulomatosis. Orbit. 2008;27(3):203-6.
14. Charles SJ, Meyer PA, Watson PG. Diagnosis and management of systemic Wegener’s granulomatosis presenting with anterior ocular inflammatory disease. Br J Ophthalmol. 1991 Apr;75(4):201-7.
