Neurofibromatosis (NF) is a genetic abnormality that affects the cell growth of neural tissue, leading to tumor growths that impact the skin, nervous system, eyes and other organs. NF is divided into two primary subgroups: neurofibromatosis type 1 (NF1), also known as von Recklinghausen or peripheral neurofibromatosis; and neurofibromatosis type 2 (NF2), also known as bilateral acoustic neurofibromatosis and central neurofibromatosis.1
NF1 and NF2 vary based on location of chromosome mutation, tumor type and location, non-tumor manifestations and management techniques; however, clinical presentations of both subtypes may overlap, making diagnosis difficult (see, “Diagnosing Neurofibromatosis.”) Other research has indicated NF can manifest in other variants including a mosaic (i.e., segmental) form, which only occurs in a localized area or organs in a linear, patchy or circumscribed area.2
Both NF1 and NF2 are acquired through an inherited autosomal dominant transmission or sporadic mutation, with presentation of NF1 more common than NF2.2 As such, members of the same family with NF may have different disease presentations from each other, as they do not always carry the same gene mutations. These can vary from complete gene deletion to insertion, stop and splicing mutations, making the exact severity of the disease difficult to predict.3 Onset of NF during childhood typically indicates a more severe progressive disease course can be expected; no sex or race predilection exists.2,4 Table 1 compares NF1 and NF2.
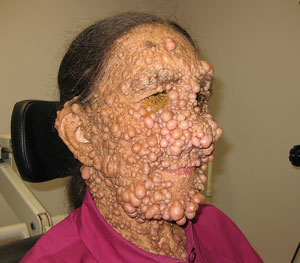 | |
| This patient’s neurofibromas are significant, but the ocular presentations, such as Lisch nodules or retinal astrocytic hamartomas, may be more difficult to identify. Photo: Al Kabat, OD. |
Neurofibromatosis 1 (NF1)
NF1 is caused by a gene mutation that affects the production of the tumor suppressor protein neurofibromin by inhibiting cell division, increasing risk of benign and malignant tumor development. Clinical features of NF1 include skin fold freckles, neurofibromas, optic pathway gliomas, Lisch nodules and the most common finding associated with the disease: café au lait spots. These hyperpigmented lesions are either seen at birth or by two years of age and increase in both size and number as the patient grows. Note, however, the presence of café au lait spots alone is not enough for a diagnosis if the child does not have a family history of the disease; he or she should instead be watched closely for additional signs of NF1 to confirm.2
In the Eye
With respect to ocular findings, the most common diagnostic criteria in NF1 patients are Lisch nodules, which appear smooth and elevated with a clear to brownish-yellow coloration on slit lamp examination. Often, these asymptomatic lesions present inferiorly and bilaterally; however, there have been reports of unilateral presentations in segmental NF patients.7 Lisch nodules rarely cause ocular complications and patients are typically asymptomatic.8
NF1 patients may also present with plexiform neurofibroma, retinal tumors and optic nerve pathway gliomas. Plexiform neurofibromas are soft swellings with indistinct borders located underneath the skin that can infiltrate the orbit and temporal regions or the eyelids. Eyelids with neurofibromas typically feel like a ‘bag of worms’ when palpitated. Orbital neurofibromas can cause strabismus or proptosis, leading to alterations in globe length, and have been associated with infantile glaucoma secondary to narrow angle.1,8 Patients younger than 10 years of age should be monitored for amblyopia, which can result from ptosis or anisometropia. Other causes of amblyopia in NF patients include lens opacity, retinal abnormalities and intracranial tumor growth.
Retinal astrocytic hamartomas are benign tumors of the retinal nerve fiber layer that can present bilaterally involving the optic nerve and posterior pole, with multiple peripheral lesions extending to the anterior retina.8,9 If the optic nerve or macula are involved, patients may exhibit decreased vision or a strabismus, while leukocoria may present if the tumor is located in the posterior pole. Glaucoma, while rare, has also been associated with anterior chamber hamartomas.9 Note, retinal astrocytic hamartomas are more commonly associated with tuberous sclerosis (TS) than NF1 and present in one of three ways: flat and semitransparent tumors in periphery with poorly defined borders; elevated, nodular, opaque white lesions with well-defined borders similar in appearance to white mulberries; or a combination of these two types.9
Diagnosing Neurofibromatosis
2. To confirm an NF2 diagnosis, bilateral vestibular schwannomas or a family history of NF2 must be present, as well as:
Other criteria include:
|
In the Brain
Optic nerve pathway gliomas (OPGs) are serious, but curable, brain tumors that arise in and around the optic nerve. Half of patients with optic nerve gliomas are NF1 patients; NF1-associated OPGs are typically less aggressive than non-associated NF1 OPGs.10 Many patients with OPGs are asymptomatic; however, OPGs may cause unilateral, bilateral or color vision loss; optic nerve pallor or atrophy; proptosis; nystagmus; or strabismus. Chiasm or adjacent brain involvement may cause endocrine and neurological symptoms.6 Typically, NF1 OPGs are stable for years and may slowly progress or spontaneously regress.11 Treatment is rarely needed.
Symptomatic OPGs typically present by age six, with most children diagnosed by age three. Often, visual acuity can be assessed by age three, color vision by age five and visual fields by age eight.6 MRI screenings are recommended every two years in patients with known OPG.12,13 Upon identification of an optic glioma, current recommendtions include ophthalmological and MRI studies quarterly for the first year, and at gradually lengthening test intervals over the next two to three years.14 Optical coherence tomography (OCT) is useful tool to monitor OPG. This noninvasive test is performed quickly and provides repeatable data. OCT will show a decrease in retinal nerve fiber layer (RNFL) when an OPG is present. MRI scanning is controversial in asymptomatic patients. The OCT detected RNFL loss in one patient before the patient had a decrease in vision.15 Providers should use the OCT if one is available.
One less common finding is choroidal abnormalities: multiple pigmented nevi and flat ill-defined choroidal hamartomas that may be light tan, yellowish-white or black in color; one to two disc diameters in size; and numbering from two to 20. They are typically located in the posterior pole and increasing in number with aging. Fluorescein angiography reveals avascular patches of hypofluorescence, similar to multiple choroidal nevi.3,16 Choroidal abnormalities with patchy appearance can also be seen using infrared monochromatic light. One study found 79% prevalence in near-infrared reflectance detected choroidal nodules in a pediatric population; however, the study population was limited, as near-infrared is not easily applied during the first years of life.16 Less common ocular findings associated with NF1 include retinal vasoproliferation tumors, prominent corneal nerves, heterochromia, café au lait spots on the eyelids, conjunctiva choristoma, optic nerve drusen, multifocal choroidal nevi, uveal melanoma and congenital or infantile glaucoma.1,3,12
Neurofibromatosis 2 (NF2)
NF2 is caused by a gene mutation that codes for a tumor suppressor protein known as merlin or schwannomin, resulting in an overproduction of schwann cells and tumor growth.5 As such, the hallmark sign of NF2 is bilateral vestibular schwannomas (VS), also known as acoustic neuromas, which present in 90% of NF2 patients and grow in size over time.2 Initial symptoms of NF2 in adults include tinnitus, hearing loss or balance dysfunction, or both, secondary to VS.
Patients may also exhibit skin tumors and physical weakness, though some may be entirely asymptomatic. Note, in younger NF2 patients, the VS may not be large enough to cause these same symptoms. Diagnosis of NF2 is typically made between 18 and 24 years of age; however, 18% of NF2 patients present before the age of 10.4
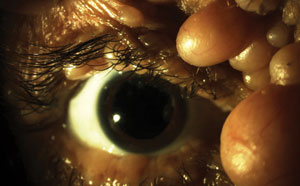 | |
| A slit lamp evaluation is an important part of monitoring the visual health of patients living with neurofibromatosis. However, eye care professionals must also be aware of the disease’s impact on retinal health, ocular motor deficits, optic atrophy and, in some cases, glaucoma risk. Photo: Al Kabat, OD. |
Juvenile posterior subcapsular lenticular opacity and peripheral cortical cataracts present bilaterally in 80% to 85% of NF2 patients and, in most cases, may be the first sign of NF2.2 Most patients are asymptomatic, but 20% experience a decrease in visual acuity; however, surgery is rarely required.4 Note, cataracts may be missed in children if a dilated examination is not performed. Obtaining a good family history can help with making an early diagnosis.
While rare in the general population, optic nerve sheath menigiomas (ONSMs) are another sign of NF2 found in up to 27% of NF2 patients.17 These benign tumors can lead to compression or vascular compromise of the optic nerve’s axons, causing progressive visual loss, color vision loss and optic atrophy or disc swelling; the triad of visual loss, optic atrophy and optociliary shunt vessels are pathognomonic for ONSMs.10 Other findings characteristic of ONSMs include proptosis and gaze evoked amaurosis at later stages. Many patients with ONSMs also have reduced ocular motility due to restriction of orbital tumor mass or paresis of oculomotor nerves caused by compression or schwannomas.10,17 ONSMs are typically unilateral and bilateral only in 5% of cases.10
Retinal hamartomas are nonmalignant focal growths that typically present unilaterally in an abnormal configuration. If they are bilateral, consider phakomatous etiologies, especially NF1, NF2, tuberous sclerosis or Gorlin syndrome.18
Combined pigment epithelium and retinal hamartomas (CPERH) are benign congential retinal tumors located typically in the posterior pole that involve the retinal pigment epithelium, neurosensory retina, retinal vessels and adjacent vitreous.19 Traction at the vitreoretinal interface may be present, producing prominent preretinal gliosis and vessel tortuosity. Vision loss can occur depending on the location and stability. CPERH has been identified in a one-year-old child who was diagnosed with NF2 six years later.20
Epiretinal membranes (ERMs) can co-occur with CPERH in 78% of cases.20 Adults presenting with ERM are typically older than 50, and etiology is typically idiopathic or secondary to ocular disease. Isolated ERMs in children are likely congenital; however they can further affect vision clarity. Bilateral ERMs may indicate a more severe NF2 phenotype.21
| Table 1. Comparison Features of NF1 and NF211 | |||
| Feature | NF1 | NF2 | |
| Frequency | 1:2,500-4,000 (more common). | 1:40,000-50,000 (rare). | |
| Inheritance | Parent with autosomal dominant condition has a 50% chance of passing onto child; 50% de novo mutations without family history. | Parent with autosomal dominant condition has a 50% chance of passing onto child; 50% de novo mutations without family history. | |
| Tumor types | Neurofibroma; glioma; malignant peripheral sheath tumor; nonlymphocytic leukemia; phenochromocytoma; Increased risk of breast cancer; gastrointestinal stromal tumors in 4% to 25% of patients. | Schwannoma; ependymoma; meningioma; glioma; malignant transformations are rare. | |
Non-tumor manifestations | Learning difficulties; skeletal dysplasia; vascular stenosis; café-au-lait macules are common; cardiovascular anomaly in 27% of patients. | Posterior subcapsular cataract/cortical wedge opacity; café au lait macules are less common. | |
| Mutation location | Chromosome 17. | Chromosome 22. | |
| Gene | Neurofibromin. | Merlin (schwannomin). | |
| Management | Take medical history; examine skin, skeleton, cardiovascular system and neurological system; perform ophthalmic evaluation biannually until age 8, then annual if stable. Gliomas that cause visual problems tend to present early in childhood; perform developmental assessment in children; perform genetic consultation; perform MRI scan for suspected cases. | Perform MRI scan of head; perform hearing evaluation; perform ophthalmic evaluation; perform cutaneous evaluation; perform genetic consultation. | |
Less common ocular findings of NF2 include ocular motor deficits due to direct or indirect compression of cranial nerves or of brainstem or cerebellum, or both. The deviation can change as the tumor grows and should be measured when examined.17 Optic atrophy may be seen in patients with papilledema, recurring papilledema or direct optic nerve compression typically secondary to increased intracranial pressure (ICP).17 Some NF patients may exhibit recurring tumor formation, resulting in recurring episodes.17 Optic atrophy can also be seen in patients presenting with ONSMs. Lid dysfunction, exophthalmous, corneal hypothesia and neurotropic keratopathy, disc edema and cranial nerve palsy have also been reported.1,2,4,22,23
Neurofibromatosis is a serious systemic disease causing tumor growth than can negatively affect the entire body. While the diagnostic criteria may not be always be met in younger patients, clinicians who are suspicious should follow them closely. In time, they may develop other NF signs that satisfy the criteria.
Both NF1 and NF2 have ocular signs that may lead to an earlier diagnosis; however, these findings are rare in the normal population, especially if diagnosed at a young age. The severity of ocular involvement and disease course can vary from patients, even members of the same family.
Patients should be referred to appropriate specialist to be monitored for any current or future complications that may arise. Patients should be educated on the fact that NF can be a progressive disease and the importance of being followed regularly by appropriate specialists, including neurosurgeons, otolaryngologists, audiologists, optometrists/ophthalmologists, neuroradiologists and geneticists.
Dr. Louprasong is a staff optometrist at the Cincinnati VA Medical Center.
Dr. Mercado is a staff optometrist at the Salisbury VA Medical Center. He is also an adjunct faculty member at the Illinois College of Optometry and the Southern College of Optometry.
1. Marks E, Adamczy D, Thomann K. Primary Eyecare in Systemic Disease. Norwalk:Appleton and Lange;1995.2. Evans D. Medial management of neurofibromatosis. Paediatr Child Health. 2001;21(10):459-465.
3. Huson S, Jones D, Beck L. Ophthalmic manifestations of neurofibromatosis. Br J Ophthalmol. 1987;71:235-238.
4. Ruggieri M, Iannetti P, Polizzi A, et al. Earliest clinical manifestations of natural history of neurofibromatosis type 2 (NF2) in childhood: a study of 24 patients. Neuropediatrics. 2005;36:21-34.
5. Antnôio JR, Goloni-Bertollo, Trídico LA. Neurofibromatosis: chronological history and current issues. An Bras Dermatol 2013;88(3):329-43.
6. Ruggieri M. The different forms of neurofibromatosis. Child’s Nerv Syst. 1999;15:295-308.
7. Adams EG, Stewart KMA, Borges OA, Darling T. Multiple unilateral Lisch nodules in the absence of other manifestations of neurofibromatosis Type 1. Case Rep Ophthal Med. Vol 2011, Article ID 854784, 2 pages.
8. Kreusel KM. Ophthalmological manifestations in VHL and NF 1: pathological and diagnostic implications. Fam Cancer 2005;4:43-47.
9. Martin K, Rossi V, Ferrucci S, Pian D. Retinal astrocytic hamartoma. Optometry. 2010;81:221-233.
10. Bosch MM, Wichmann WW, Boltshauser E, Landau K. Optic nerve sheath meningiomas in patients with Neurofibromatosis Ttype 2. Arch Ophthalmol. 2006;124:379-385.
11. Korf B. Malignancy in neurofibromatosis Type 1. Oncologist. 2000;5:477-485.
12. Ferner RE, Huson, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis type1. J Med Genet. 2007;44:81-88.
13. Segal L, Darvish-Zargar M, Dilenge ME, et al. Optic pathway gliomas in patients with neurofibromatosis type 1: Follow-up of 44 patients. J AAPOS 2010;14:155-158.
14. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834-43.
15. Parrozzani R, Clementi M, Kotsaffio O, Miglionico G, Trevisson E, Orlando G, Pilotto E, Midena E. Optical Coherence Tomography in Diagnosis of Optic Pathway Gliomas. Invest Ophthalmol Vis Sci. 2013 Dec 17;54(13):8112-8.
16. Makino S, Tampo H, Arai Y, Obata H. Correlations between choroidal abnormalities, Lisch nodules, and age in patients with neurofibromatosis type 1. Clin Ophthalmol. 2014;8:165-168.
17. Bosch MM, Boltshauser E, Harpes P, Landau K. Ophthalmologic Findings and Long-Term Course in Patients with Neurofibromatosis Type 2. Am J Ophthalmol. 2006;141(6):1068-1077.
18. Grant EA, Trzupek KM, Reiss J, et al. Combined retinal hamartomas leading to the diagnosis of neurofibromatosis type 2. Ophthalmic Genet. 2008;29:133-138.
19. Vianna RNG, Pacheco DF, Vasconcelos MM, De Laey JJ. Combined hamartoma of the retina and retinal pigment epithelium associated with neurofibromatosis type-1. Int Ophthalmol. 2002;24:63-66.
20. Landau K, Yasargil GM. Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol. 1993;77:646-649.
21. Siwiec-Proscinska J, Gotz-Wieckowska A, Pawlak, Kociecki J. Epiretinal membrane and cataract in a 5-year-old boy with the suspicion of neurofibromatosis type 2. Cent Eur J Med. 2013;8(1):80-83.
22. Ragge NK. Clinical and genetic patterns of neurofibromatosis 1 and 2. Br J Ophthalmol. 1993;77:662-672.
23. Ragge NK, Baser ME, Riccardi VM, Falk RE. The ocular presentation of neurofibromatosis 2. Eye. 1997;11:12-18.
