Just because a disease is rare doesnt mean it wont end up in your chair. As a result, you must be knowledgeable of the rare systemic conditions that have ocular manifestations so you are not misled to make a diagnosis based solely on the appearance of a particular corneal or retinal finding. This article discusses three such rare systemic diseases: Fabrys disease, Churg- Strauss syndrome and Susacs syndrome (see Diseases at a Glance, below.), and stresses the importance of looking past the eye and considering the patients entire body. Diseases at a Glance
Disease
Etiology
Sex Predominance
Treatment
Fabrys
Sex-linked recessive
metabolic disorder M>F
Agalsidase
beta IV
infusion q2 weeks
Churg-Strauss
ANCA-positive vasculitis
M>F
Steroid + cytotoxic agent
Susacs
Unknown
F>M
steroid
Fabry"s Disease
Fabrys disease, also known as Anderson-Fabry disease or angio-keratoma corporis diffusum, is an X-linked recessive metabolic disorder occurring in 1 in 40,000 males.1 It is the second most prevalent metabolic storage disease of humans, affecting all ethnicities, with an incidence of 1 in 117,000 in the general population.2,3 Men with Fabrys disease (hemizygotes) are predominantly affected, but many female carriers (heterozygotes) have similar clinical involvement.4,5 Patients as young as five years of age may present with early intermittent symptoms (to be discussed below). But, because the disease often goes unrecognized by physiciansbecause it causes many varied symptoms affecting different body parts, the mean age of diagnosis for the illness is around 29 years.3,6
Fabrys disease is caused by an inborn error of glycosphinogolipid metabolism, which results from the defective activity of the lysosomal hydrolase, a-galactosidase A (a-GAL). This deficiency of the enzyme a-GAL leads to the accumulation of glycosphingolipids, particularly globotriaosylceramide (GL-3), within vascular endothelium, smooth muscle cells and visceral organs.7 GL-3 can be found in renal epithelium, myocardium, dorsal root ganglia, the brain and the autonomic nervous system.8
The classic systemic findings begin in childhood or adolescence and include intermittent episodes of burning and aching pains in the hands and feet (acroparesthesias); abdominal pain, hypohidrosis or anhidrosis (impaired ability to perspire); heat, cold and exercise intolerance; episodic fevers; and dark reddish-purple, non-blanching lesions (angiokeratomas)usually seen in a bathing suit distribution around the umbilicus, buttocks, groin and upper thighs (though these lesions may also involve the eyelids).1,7,8
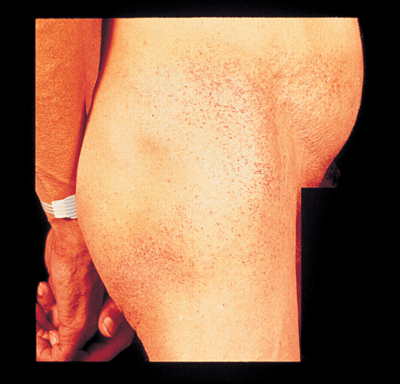 |
| The classic systemic findings of Fabrys disease include dark reddish-purple, non-blanching lesions (angiokeratomas) usually seen in a bathing suit distribution around the umbilicus, buttocks, groin and upper thighs, as shown above. Still, these lesions may also involve the eyelids. |
Gastrointestinal symptoms in patients who have Fabrys disease include episodic diarrhea, post-prandial bloating and pain, and weight loss.7 Bronchitis, dyspnea and wheezing are not uncommon.1 Various cerebrovascular events, such as stroke, transient ischemic attacks, hemiparesis, vertigo/dizziness and dementia can also occur.1,7
The most common and first ocular sign of Fabrys disease: bilateral corneal diffuse yellow epithelial haziness that gradually becomes concentrated into dense rays that radiate from the center of the cornea into dense bronze to cream-colored streaks arranged in a vortex or star-shaped pattern (whorl-like or verticillata).8,9 These opacities appear to be in the subepithelial or Bowmans layer of the cornea.8 Corneal involvement occurs in over 90% of these patients, and, in one study, this involvement was present in all female carriers over the age of 10 years.10,11 In my experience, I have found that these opacities are best seen by transillumination and can mimic similar findings that may occur in patients who take amiodarone (an antiarrhythmic), chloroquine (used to prevent and treat malaria and liver disease), or chlorpromazine (used to treat psychotic disorders, such as delusions).
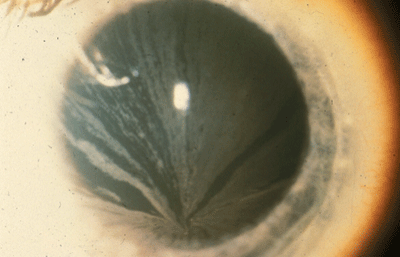 |
| In Fabrys disease, bilateral corneal diffuse yellow epithelial haziness eventually becomes dense bronze to cream-colored streaks arranged in a vortex or star-shaped pattern (whorl-like or verticillata). |
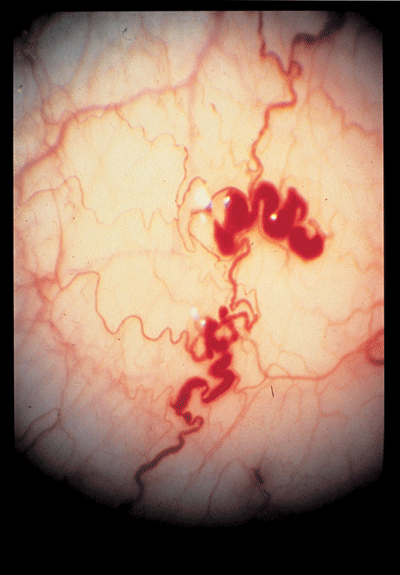 |
| The small vessels of the conjunctivea in Fabry"s disease often show aneurysmal dilations, tortuosity, and kinking. |
Retinal vascular changes are more prevalent in hemizygous males (70% vs. 25% in heterozygous females).8 These vascular changes may present as segmental sausage-like dilation of the veins or corkscrew-like diffuse tortuosity.8 Other retinal findings include hemorrhages, hypertensive retinopathy, perimacular edema and central retinal artery occlusion.8,9,11,12
Ocular abnormalities may also occur secondary to neurologic disease and include nystagmus, diplopia from extraocular muscle palsies or internuclear ophthalmoplegia, disc edema, anterior ischemic optic neuropathy, hemianopsia and pupil abnormalities.8,9,11,12 (See Ocular Signs and Symptoms of Fabrys Disease," below.)
|
Ocular Signs and Symptons of Fabry"s Disease | |
|
Eyelid and Periorbita |
Edema |
|
Conjunctiva |
Aneurysmal dilation, tortuosity |
|
Cornea |
Diffuse yellow epithelial |
|
Lens |
Anterior capsular propeller |
|
Retina |
Sausage-like dilation of veins |
|
Neurologic |
Nystagmus |
|
Autonomic |
Decreased tear production |
Aside from the clinical evaluation and a positive family history of the disease, laboratory findings of a marked decrease in activity of the enzyme a-GAL help confirm the diagnosis of Fabrys disease. In 2003, the FDA approved the re-combinant human a-GAL enzyme Fabrazyme (agalsidase beta, Genzyme) as a replacement therapy in Fabrys disease patients. This drug has been shown to reduce GL-3 deposition and slow the progression of the disease process.14 It is administered as an intravenous in-fusion every two weeks.14 If the cataracts are affecting the patients vision, they should be surgically removed. Treat the patients dry eye with ocular lubricants and/or punctal plugs. Unfortunately, the other aforementioned ocular complications cannot be treated.
Churg-Strauss Syndrome
Churg-Strauss syndrome (CSS), also known as allergic granulomatous angiitis, is a distinct vasculitic syndrome belonging to the pauci-immune antineutrophil cytoplasmic antibody (ANCA) positive vasculitides.15 It is an uncommon condition characterized by asthma, eosino-philia and a recurrent necrotizing vasculitis.15,16
CSS affects men twice as often as women, and the age of onset varies from 15 to 70 years, with a mean age of about 40 years.17 The cause is unknown. The distinct nature of CSS distinguishes it from polyarteritis nodosa, a disease of small-sized and medium-sized arteries.15
Several stages of CSS can usually be identified:
Prodromal stage. This can persist for years and is manifested by symptoms of allergic disease with rhinitis, nasal polyps, bronchitis with shortness of breath, fever and asthma.16,18
Infiltration stage. This occurs when blood eosinophil levels become elevated, and the skin, lungs and gastrointestinal tract are infiltrated by these cells (eosinophils).18
Vasculitis stage. This is generalized (symptoms affect many different organ systems) and accompanied by weight loss, malaise, fever, arthralgias, myalgias and cutaneous vasculitis. More severe manifestations occur with persistent perivascular eosinophilic infiltration. These manifestations include cardiac and renal failure, convulsions and coma.18
Ocular involvement of CSS is uncommon, although eosinophilic granulomas may develop on the eyelids and conjunctiva.16,19 Similar granulomatous lesions can cause episcleritis, scleritis and panuveitis.15 Marginal corneal ulceration can also occur.16 Neuro-ophthalmologic findings include episodes of transient monocular visual loss, retinal ischemia, branch and central retinal artery occlusion, ischemic optic neuropathy and various cranial neuropathies including ocular motor nerve palsies.20
If the disease is diagnosed early, the outlook is relatively good, meaning treatment may halt organ damage or slow the disease process. Treatment consists of a combination of oral or intravenous steroids with a cytotoxic agent, such as oral cyclophosphamide.17,18 If left untreated, CSS is frequently fatal.16 An eyelid or conjunctival biopsy of involved tissue helps identify any eosinophilic granulomas and therefore helps in the diagnosis. Laboratory tests in a complete uveitis work-up should also be done to verify the diagnosis if these tests show eosinophilia > 10% or > 400/ml in peripheral blood and presence of antineutrophil cytoplasmic antibodies.
Susac"s Syndrome
Susacs syndrome was first described in 1979 and is defined by a triad of multiple branch retinal arterial occlusions, brain microangiopathy causing encephalopathy, and sensorineural hearing loss.21 This condition usually occurs in young women but can affect 15% to 20% of men.22,23 The age range for both men and women is from eight to 59 years with a mean age of 28.1 years and a median age of 29 years.22 No racial trend is noted.22 Its etiology is not known.
The retinopathy seen in Susacs syndrome is characterized by multiple peripheral retinal arteriolar branch occlusions that can be seen on funduscopy or retinal fluorescein angiography. The occlusions may be quite extensive or very subtle.22,24 Aside from the retinal arteriolar occlusions and their accompanying vessel narrowing, boxcar segmentation of the blood column may be seen beyond the point of occlusion in the affected arterioles, pointing to thrombosis as a possible mechanism for the occlusions.25 Iritis and vitritis are not seen, and there is usually no periarteriolar inflammation or sheathing.25,26
Retinal fluorescein angiography typically shows retinal arterial wall hyperfluorescence.27 The hyperfluoresence serves as an indicator of active disease and can precede the actual arterial occlusion.22 Vessel wall hyperfluoresence may be seen at sites distant from and unassociated with those of retinal arterial occlusion. Fluorescein angiography should always be performed on patients suspected of having Susacs syndrome, as the more subtle branch arterial occlusions may only be detected by doing this procedure. Other signs seen with fluorescein angiography: arterial constriction, leakage and perivascular sheathing.22,27
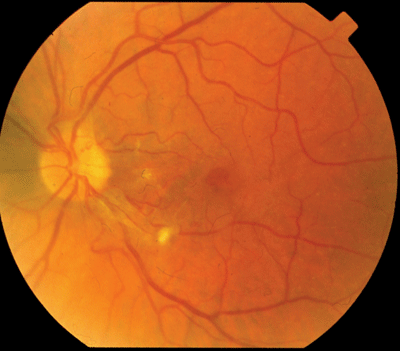 |
| The retinopathy seen in Susacs syndrome is characterized by multiple peripheral retinal arteriolar branch occlusions that can be seen on funduscopy or retinal fluorescein angiography. |
Hearing loss in these patients is often acute, unilateral or bilateral, asymmetrical and associated with tinnitus, vertigo, vomiting, nausea and unsteady gait.27,23 The vertigo may be accompanied by nystagmus.27 The progression of the hearing loss is variable during the active phase of the illness. Some patients experience rapid progression and deafness, whereas others have significant improvement of previously impaired cochlear function on one side and subsequent involvement contralaterally.29
The encephalopathy may be acute or subacute and is frequently seen with psychiatric features of personality change and bizarre behavior.29 Common neuropsychiatric signs and symptoms include global brain microangiopathy, memory loss, headache (often with migrainous features), impaired cognition, ataxia, dysarthria, hemiparesis, hemisensory changes and brain atrophy.22,29
Magnetic resonance imaging (MRI) is the neuro-imaging technique of choice for identifying the etiology of the encephalopathy. The MRI findings show multiple small foci of abnormality. These appear both in the gray and white matter of the brain as hyperintensities.29 These lesions, which enhance on contrast dye (gadolinium) injection, are ischemic infarctions.30
The disease has a spontaneous remitting-relapsing course. The neuropsychiatric symptoms may resolve completely, but any hearing loss or visual changes usually do not improve. The mainstay of treatment is a minimum of 1 mg/kg oral prednisone (up to 80mg daily) with slow taper over two weeks, preceded by three days of intravenous methylprednisolone in severe cases. A total of 87% to 91% of patients have a response to prednisone treatment.22
Since retinal changes usually mirror a neurologic brain disease, weekly funduscopic exams are recommended for the first month, followed by monthly exams. Disease activity can also be assessed by monthly or quarterly MRIs and hearing evaluations. New lesions seen on MRI will enhance brightly than older lesions, indicating disease progression.29,31
Awareness of the aforementioned rare systemic diseases that have ocular manifestations will ensure that you ask patients who present with these particular corneal or retinal findings the proper questions during the history portion of their exam. This is crucial to help you make the correct diagnosis, and prescribe the appropriate treatment. Again, just because its rare, that doesnt mean it wont end up in your chair.
Dr. Skorin is the staff osteopathic ophthalmologist at Albert Lea Eye ClinicMayo Health System, Albert Lea, Minn.
1. Desnick RJ, Ioannou YA, Eng CM. Alpha-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th ed. New York: McGraw Hill, 2001; 3733-74.
2. Brady RO, Schiffmann R. Clinical features of and recent advances in therapy for Fabry disease. JAMA 2000 Dec 6; 284(21):2771-5. Erratum in: JAMA 2001 Jan 10;285(2): 169.
3. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999 Jan 20;281(3): 249-54.
4. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001 Nov;38(11):769-75.
5. Wendrick K, Whybra C, Ries M, et al. Neurological manifestation of Fabry disease in females. Contrib Nephrol 2001; 136:241-244.
6. Morgan SH, Crawfurd MA. Anderson-Fabry disease. BMJ 1988 Oct 8;297(6653):872-3. Review.
7. Genzyme Corp. Fabry Disease. Disease Monograph. Cambridge, MA, 2003:1-20.
8. Ayazifar M, Snady-McCoy L. Abnormal-appearing retinal vessels. In: Steidl SM, Hartnett ME (eds). Clinical Pathways in Vitreoretinal Disease. New York: Thieme, 2003:183-202.
9. Ostler HB, Maibach HI, Hoke AW, Schwab IR. Diseases of the Eye and Skin: A Color Atlas. Philadelphia: Lippincott Williams & Wilkins, 2004:158-159.
10. Franceschetti A. Fabrys disease: ocular manifestations. In: Bergsma D, Bron AJ, Cotlier E (eds). The Eye and Inborn Errors in Metabolism. Vol. 12, No. 3. New York: AR Liss Co., 1976: 195-208.
11. Sher NA, Letzen RD, Desnick RJ. The ocular manifestations in Fabrys disease. Arch Ophthalmol 1979 Apr;97(4): 671-6.
12. Repka MX. Degenerative and metabolic disease in infants and children. In: Miller NR, Newman NJ (eds). Walsh & Hoyts Clinical Neuro-Ophthalmology, 6th ed., Vol. 3. Phila-delphia: Lippincott Williams & Wilkins, 2005; 2469-2511.
13. Cable WJL, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology 1982; May;32(5): 498-502.
14. Genzyme Corp. Fabrazyme (agalsidase beta) package insert. Cambridge, MA, 2004.
15. Messmer EM, Foster CS. Vasculitic peripheral ulcerative keratitis. Surv Ophthalmol 1999 Mar-Apr;43(5):379-96.
16. Kaufman HE, Barron BA, McDonald MB, Kaufman SC. Companion Handbook to the Cornea, 2nd ed. Boston: Butter-worth Heinemann, 2000: 571-572.
17. Calabrese LH, Duna G. Vasculitis associated with antineurophil cytoplasmic antibody. In: Ruddy S, Harris ED, Sledge CB, et al. (eds). Kelleys Textbook of Rheumatology, 6th ed. Philadelphia: WB Saunders, 2001:1165-1184.
18. Roecken M, Grevers G, Burgdorf W. Color Atlas of Allergic Diseases. New York: Thieme, 2003:142-143.
19. Shields CL, Shields JA, Rozanski TI. Conjunctival involvement in Churg-Strauss syndrome. Am J Ophthalmol 1986 Nov 15;102(5):601-5.
20. Weinstein JM, Chui H, Lane S, et al. Churg-Strauss syndrome (allergic granulomatous angiitis): Neuro-ophthalmologic manifestations. Arch Ophthalmol 1983 Aug;101(8): 1217-20.
21. Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology 1979 Mar;29(3):313-6.
22. OHalloran HS, Pearson PA, Lee WB, et al. Microangio-pathy of the brain, retina, and cochlea (Susac Syndrome). Ophthalmology 1998 Jun;105(6):1038-44.
23. Petty GW, Engel AG, Younge BR, et al. Retinocochleo-cerebral vasculopathy. Medicine (Baltimore). 1998 Jan;77(1):12-40. Review.
24. Susac JO. Susacs syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology 1994 Apr;44: 591-593.
25. Pfaffenbach DD, Hollenhorst RW. Microangiopathy of the retinal arterioles. JAMA 1973 Jul 30;225(5):480-3.
26. Bogousslavsky J, Gaio JM, Caplan LR, et al. Encephalopathy, deafness and blindness in young women: A distinct retinocochleocerebral arteriolopathy? J Neurol Neurosurg Psychiatry 1989 Jan;52(1):43-6.
27. Papo T, Biousse V, Lehoang P, et al. Susac syndrome. Medicine (Baltimore) 1998; 77: 3-11.
28. Monteiro ML, Swanson RA, Coppeto JR, et al. A micro-angiopathic syndrome of enceph-alopathy, hearing loss, and retinal arteriolar occlusions. Neurology 1985 Aug;35(8):1113-21.
29. Do TH, Fisch C, Evoy F. Susac syndrome: Report of four cases and review of the literature. Am J Neuroradiol 2004 Mar;25(3):382-8.
30. Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susacs syndrome. Neurology 2003 Dec; 61:1783-87.
31. White ML, Zhang Y, Smoker WRK. Evolution of lesions in Susac syndrome at serial MR imaging with diffusion-weighted imaging and apparent diffusion coefficient values. Am J Neuroradiol 2004 May;25(5):706-13.
