A mother (“Betty”) and son (“John”) presented with significantly different cases of Best’s disease. The 53-year-old mother had bilateral vitelliform lesions and subretinal fluid lesions. The 23-year-old son had no macular anomaly and an unaided monocular visual acuity of 20/15. Both mother and son had otherwise unremarkable anterior and posterior ocular findings. A comparison of their courses showed how varied this disease can be.
Here are some critical considerations to keep in mind when diagnosing and managing these patients.
Initial Presentations
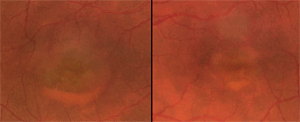
Betty had bilateral vitelliform lesions, captured on retinal fundus photography.
When Betty came to our office, her best-corrected visual acuity through hyperopic astigmatic corrective lenses measured 20/40 O.D. and 20/25 O.S. A central inferior visual field depression was detected in her right eye, but no defects were found in her left eye. She complained of a recent onset of centrally scattered scotomas. A yellow circular spot had been growing in the vision of her left eye during the previous week. She also had a long-standing central scotoma in the right eye, accompanied by decreased visual acuity in both eyes. She reported no other ocular or systemic symptoms or complications.
No issues were identified during an examination of Betty’s son, John.
Diagnostics
A battery of tests was performed to confirm the diagnosis of Best’s disease and establish a hereditary etiology in the mother and son. We completed electrooculography (EOG), electroretinography (ERG), Humphrey visual fields (24-2 and 30-2 with foveal threshold), optical coherence tomography (OCT) and fundus photography. In addition, we ordered bilateral fundus fluorescein angiography (FFA) and Amsler grid testing for Betty.
We chose EOG because patients with Best’s disease exhibit lower amplitude base values and Arden ratios (light peak/dark trough ratio) that are less than 1.5, below a ratio of more than 1.8 in patients without the disease. A normal ERG result helps confirm Best’s because the disease disrupts retinal pigment epithelium (RPE) depolarization, not the rod and cone receptors evaluated during ERG.1-7
Besides electrophysiologic or fundus observations, a diagnosis of Best’s disease can be determined with genetic testing. Molecular genetic isolation, such as polymerase chain reaction (PCR), may identify a defect of VMD2, the gene coding for the basolateral Ca2+/Cl- channel protein bestrophin in the RPE.
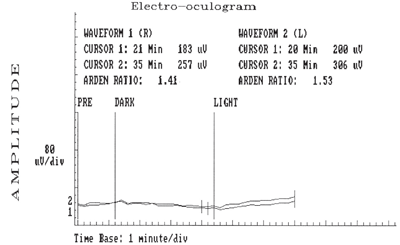
Electrooculopathy on 53-year-old Betty (above) and her 23-year-old son (below) revealed the presence of bilateral, flat, light peak to dark trough (signifying an abnormal standing potential of both eyes), bilateral Arden ratios of less than 1.5 and base amplitudes of 80mV/division.
Although an autosomal dominant inheritance has been established, a mutation of VMD2 may present de novo. Normal or minimally affected maculas are found in 5% of patients with VMD2 mutations.8,9
A mutation of bestrophin creates an imbalance in the conduction of anions through the RPE membrane, as seen on EOG, which exhibits a reduced or flat light rise and a lower base value throughout all stages of the disease. Thus, EOG findings do not correspond to visual acuity.
Diagnostic Findings
The following results were found in Betty and John:
• EOGs revealed bilateral flat light peak to dark trough (signifying an abnormal standing potential of both eyes), bilateral Arden ratios of less than 1.5 and base amplitudes of 80mV/division• ERG findings that showed normal a- and b-waves in both eyes
• No field defects in either eye, as measured by 24-2 visual fields.
Dissimilarities were detected in 30-2 visual fields with foveal fixation. An inferior central scotoma was found in Betty’s right eye. The scotoma indicated a more advanced stage of Best’s, which correlated objectively to more elevated macular cystic lesions viewable bilaterally through OCT and correlated subjectively to inferior scotomas and metamorphopsia revealed on the Amsler grid.
As I mentioned earlier, Betty had bilateral vitelliform lesions, captured through retinal fundus photography. Her right eye’s lesion was more elevated and larger in diameter than that of her left eye (one disc diameter vs. two thirds).
A positive Watzke-Allen slit beam test sign was found in her right eye only. The FFA revealed bilateral central cystic retinopathy without neovascular leakage and an incomplete ring of parafoveal blockage.
Conversely, John had no ocular anomalies measurable through visual field testing, OCT, or retinal fundus observation. He presented as a carrier of Best’s disease.
Pathophysiology
Best’s disease is a rare, incurable, autosomal dominant, slowly developing vitelliform (yolk-like) macular dystrophy associated with central visual disturbance and loss. The disease is limited to macular anomalies, excluding systemic associations. There is no gender predisposition.
Onset is usually three to 15 years of age, although it is typically not detected until the third to fourth decade of life, when symptoms develop. The disease is most often found in Caucasian individuals of European descent, but also with less frequency in populations of African and Hispanic descent.1,8,12
As we saw in Betty and John, the clinical presentation varies, ranging from reduced visual acuity and macular lesions to no symptoms or retinal anomalies. Patients should be advised to seek genetic counseling. Since the disease is autosomal dominant, the probability of an affected individual’s offspring inheriting the disease is 0.5, or 50%.
Because bestrophin function is faulty in these patients, aggregates of yellow lipofuscin derived internally from outer segments of the PR and externally from the RPE accumulate beneath the sensory retina, causing a characteristic yolk-like appearance.8,13
Staging
Best’s disease progresses through four recognized stages:
• Stage I is previtelliform, and marks the initial development of retinal defects.
• Stage II is the vitelliform stage, characterized by the early egg yolk (typically one half to one disc diameter in size) and, later, scrambled egg appearances. Patients may retain good visual acuity until the scrambled egg stage. Cystic lesions are formed in Stage II and often correspond to visual disturbances. Betty appeared to be entering this stage.
• Stage III presents layering of the fluid subretinal material, called a pseudohypopyon, and partial reabsorption of the cysts.
• Stage IV corresponds to greater hyperpigmentation, possible neovascularization and fibrous scarring in the atrophic stage. Late-stage is seen with a deterioration of central vision to 20/400 and typically occurs after 40 years of age.1
Progression through these stages is slow and variable, and may not correspond directly to visual function.
Confounding Factors
Although the diagnoses in these patients were rather straightforward, other cases may not be. Best’s disease can be mistaken for a number of macular anomalies, most commonly adult-onset vitelliform dystrophy (AVMD), age-related macular degeneration (AMD) and central serous chorioretinopathy (CSR).14-17 All three of these conditions can present with central visual disturbance, macular cystic lesions, and macular lipofuscin-like deposits. Below are factors to keep in mind:
• AVMD presents with macular vitelliform lesions that are similar in appearance and structure to those of Best’s. However, AVMD is a mutation of the RDS peripherin gene, not a mutation of the VMD2 gene.14,18 AVMD has a later onset than Best’s and is associated with normal EOG findings. AVMD also does not progress through the four stages of Best’s.
• AMD also has a later onset than Best’s, produces normal EOG findings and has the potential to progress through two stages––dry and wet. The visual prognosis is poor and foveal leakage is characteristic in the wet stage. The prognosis seen in patients with Best’s and AVMD is generally good.
• CSR typically presents unilaterally in males 25 to 50 years of age and is associated with conditions such as Type A personality, high stress situations, systemic hypertension, hypercortisolism or sleep apnea.19,12 Although leakage from the choriocapillaris is seen with serous detachment, the prognosis is good. Spontaneous resolution is commonly seen within six months, especially when stress factors are reduced.
Treatment Options
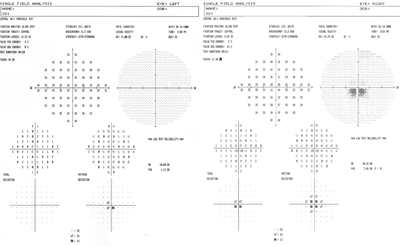
A Humphrey visual field on 53-year-old Betty identified an inferior central scotoma, indicating that she had a more advanced stage of Best’s than her son.
Currently, treatments for Best’s disease are limited. Cystic lesions may subside after administration of oral acetazolamide. Subfoveal choroidal neovascularization (CNV), which may be detected through close monitoring of the disease in its later stages, should be treated promptly to preserve visual acuity. Close monitoring of the disease in its later stages helps detect subfoveal choroidal neovascularization, which should be treated promptly to preserve visual acuity.
The treatment options we considered for Betty included photodynamic therapy (PDT), using verteporfin and argon laser photocoagulation.20 We also considered a 1-mg intravitreal injection of bevacizumab (Avastin, Genentech), which has been shown to lead to CNV regression and to restore visual acuity.21,22
Betty and John were urged to avoid smoking, ocular ultraviolet exposure and caffeine intake, which are hypothesized to increase the progression of neovascularization.
Additionally, it has been suggested that a diet similar to the one proposed through in AREDS 2 for patients with AMD may slow the progression of visual loss in Best’s. For Betty, as central acuity decreases, vision rehabilitation may also become important.
Monitoring and Follow-up
Ancillary tests were planned to monitor progression and confirm stages of disease in Betty and John. A multifocal ERG can reveal reduced amplitudes in the parafoveal concentric area. An OCT scan of the macula can be used to view the optically clear cystic elevations and hyper-reflective lipofuscin deposits in Betty’s eyes.
Macular thickness measured with OCT correlates with visual acuity; thinner retinas have poorer acuity.12,23-25 Autofluorescent photography is a highly sensitive method for measuring changes in the highly fluorescent lipofuscin deposits.1 Additionally, an FFA can demonstrate hyperfluorescence with areas of blockage due to RPE disruption and, if present, neovascular leakage. Humphrey visual fields or an Amsler grid can reveal central disturbances.
Continuing Management
Best’s disease is a complex and potentially beguiling condition that raises diagnostic challenges and defies a cookbook approach to management. Treatments are also not very promising. Counseling patients on lifestyle habits and anticipation of future problems through the use of genetic evaluation are important considerations, no matter how significant or modest the symptoms may be. John will also benefit from close follow-up in the years ahead.
1. Renner AB, Tillack H, Kraus H, et al. Late Onset is Common in Best Macular Dystrophy Associated with VMD2 Gene Mutations. Ophthalmology 2005;112:586-92.
2. Thorburn W, Nordström S. EOG in a large family with hereditary macular degeneration. (Best’s vitelliform macular dystrophy) identification of gene carriers. Acta Ophthal (Copenh) 1978;56(3):455-64.
3. Walter P, Brunner R, Heimann K. Atypical presentations of Best’s vitelliform macular degeneration: clinical findings in seven cases. Ger J Ophthal 994;3(6):440-4.
4. Alanko HI. Clinical electro-oculography. Acta Ophthal Suppl 1984;161:139-48.
5. Shinomiya K, Shiota H, Ohgi Y, et al. Analysis of the characteristics of electrooculogram applied a battery model to the eyeball. Biom and Pharm Eng 2006;428-31.
6. Wajima R, Chater SB, Katsumi O, et al. Correlating visual acuity and electrooculogram recordings in Best’s disease. Ophthalmologica 1993;207(4):174-81.
7. Bonanomi MTBC, Maia OO Jr, Lower LMT, Nakashima T. Descolamento viteliforme macular associado a drusas da lâmina basal: relato de caso. Arq Bras Oftalmol 2006;69(2): 269-72.
8. Mullins RF, Kean T, Heffron E, et al. Late Development of vitelliform lesions and flecks in a patient with Best disease. Arch Ophthalmol 2005;123:1588-94.
9. Marchant D, Yu K, Bigot K, et al. New VMD2 gene mutations identified in patients affected by Best Vitelliform macular dystrophy. J Med Genet 2007;44:e70.
10. Apushkin MA, Fishman GA, Taylor CM, Stone EM. Novel de novo mutation in a patient with Best macular dystrophy. Arch Ophthalmol 2006;124:887-9.
11. Blodi C, Stone E. Best’s vitelliform dystrophy. Ophthal Paediatr Genet 1990;11:49-59.
12. Haimovici R, Koh S, Gagnon DR, et al. Risk factors for central serous chorioretinopathy: a case-control study. Ophthalmology. 2004;111(2):244-9.
13. Spaide RF, Noble K, Morgan A, Freund B. Vitelliform macular dystrophy. Ophthalmology 2006; 113:1392-400.
14. Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet 2003;40:641-50.
15. Do P, Ferrucci S. Adult-onset foveomacular vitelliform dystrophy. Optometry 2006;77:156-66.
16. Arnold JJ, Sarks JP, Killingsworth MC, et al. Adult vitelliform macular degeneration: a clinicopathological study. Eye 2003;17:717-26.
17. Spaide RF. Deposition of yellow submacular material in central serous chorioretinopathy resembling adult-onset foveomacular vitelliform dystrophy. J Ret Vit Dis 2004; 24(2):301-4.
18. Renner AB, Tillack H, Kraus H, et al. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina 2004;24:929-39.
19. Loo RH, Scott IU, Flynn HW Jr, et al. Factors associated with reduced visual acuity during long-term follow-up of patients with idiopathic central serous chorioretinopathy. Retina 2002; 22(1):19-24.
20. Andrade RE, Farah ME, Cardillo JA, et al. Optical coherence tomography in choroidal neovascular membrane associated with Best’s vitelliform dystrophy. Acta Ophthalmol Scand 2002;80:216-218.
21. Leu J, Schrage NF, Degenring RF. Choroidal neovascularization secondary to Best’s disease in a 13-year old boy treated by intravitreal bevacizumab. Graefe’s Arch Clin Exp Ophthalmol 2007; 245:1723-5.
22. Chan WM, Lai TY, Liu DT, Lam DS. Intravitreal bevacizumab (Avastin) for choroidal neovascularization secondary to central serous chorioretinopathy, secondary to punctate inner choroidopathy, or of idiopathic origin. Am J Ophthalmol 2007;143(6):977-98.
23. Vedantham V, Ramasamy K. Optical coherence tomography in Best’s disease: an observational case report. Am J Ophthalmol 2005;139(2):351-3.
24. Men G, Batıoğlu F, Özkan SS, et al. Best’s vitelliform macular dystrophy with pseudohypopyon: An optical coherence tomography study. Am J Ophthalmol 2004;137(5):963-4.
25. Arora R, Das S, Shroff D, et al. In vivo microscopy of Best’s vitelliform macular dystrophy: Optical coherence tomography study of combined stage III and IV lesions. Clin Exp Ophthalmol 2007;35:287-8.
