 |
An 11-year-old male with complaints of gradually blurring vision in both eyes was referred to our office after a year of experiencing symptoms. The accompanying parent noted that the child had a normal birth, growth and development. His pertinent medical history was remarkable for asthma. Family history was remarkable for unknown vision loss suffered by his maternal grandfather and a maternal cousin. Otherwise, the boy’s ocular history was noncontributory.
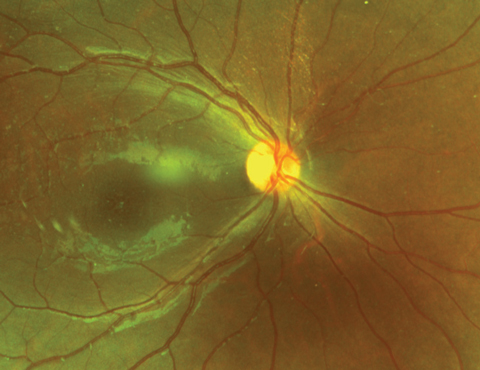 |
| Fig. 1 This fundus photograph shows an 11-year-old patient’s right eye. |
Examination
Upon examination, visual acuities with his current spectacle correction were 20/70 OD and 20/100 OS with no improvement with pinhole OU. Pupils were equally round and reactive to light. There was no afferent defect. Confrontation visual fields were full to careful finger counting in both eyes. Extraocular motility testing was normal. Anterior segments of both the right and left eye were unremarkable. Applanation tonometry measured 19mm Hg OD and 17mm Hg OS.
Fundus examination of the right eye revealed normal vitreous and optic nerve, with tortuous vessels, and a poor foveal light reflex. Other changes were noted in fundus images (Figure 1). Similar and more extensive finding were seen in the left eye (Figure 2). A fluorescein angiogram (FA) and OCT were obtained and available for review (Figures 3-6).
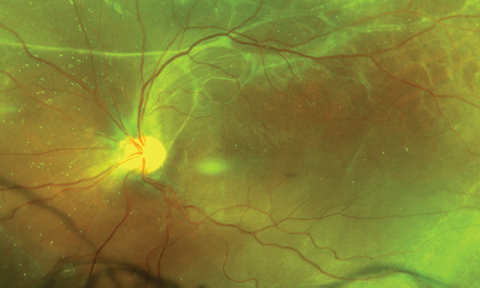 |
| Fig. 2. Pertinent findings were also discovered in his left eye, shown below. |
Take the Retina Quiz
1. What is the most likely diagnosis?
a. Retinitis pigmentosa.
b. Juvenile X-linked retinoschisis.
c. Cystoid macular edema.
d. Goldmann-Favre vitreoretinal degeneration.
2. What is the appropriate interpretation of the OCT imaging of the right eye as seen in Figure 5?
a. IS/OS disruption.
b. Epiretinal membrane (ERM).
c. Cystic changes.
d. Vitreomacular traction.
3. What is the inheritance pattern of this condition?
a. Autosomal dominant.
b. Autosomal recessive.
c. X-linked dominant.
d. X-linked recessive.
4. Aside from surgical intervention, what else is warranted in the management of this patient?
a. Genetic testing of patient and family members.
b. Yearly observation.
c. Anti-VEGF injection.
d. Vitamin A supplementation.
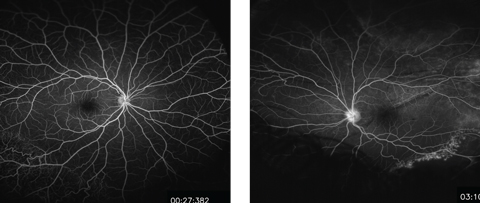 |
| Figs. 3 and 4. These midphase FA images show the patient’s right (at left) and left eyes. Do they reveal any pathology? Click image to enlarge. |
Diagnosis
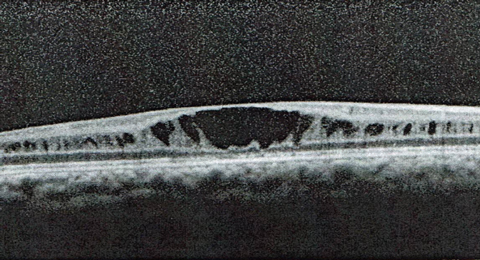
The macula of the right eye showed cystic spaces within the sensory retina. This was confirmed using OCT, where macular cysts can be seen (Figure 5). In the left eye, there was an obvious macular detachment, which was also confirmed with OCT imaging (Figure 6). The right eye showed prominent white without pressure and fibrotic bands extending into the vitreous. A peripheral retinoschisis was present inferotemporally. The fundus exam of the left eye revealed pigmented cells in the vitreous, retinal folds and fibrotic bands extending superiorly and superonasally. The peripheral retina was superiorly detached.
The FA revealed vascular abnormalities in the patient’s inferotemporal peripheral retina in the right eye and inferonasal peripheral retina in left eye (Figures 3 and 4). After careful review of the patient’s familial ocular history, ancillary testing and clinical presentation, the young boy was diagnosed with juvenile X-linked retinoschisis (JXRS).
 |
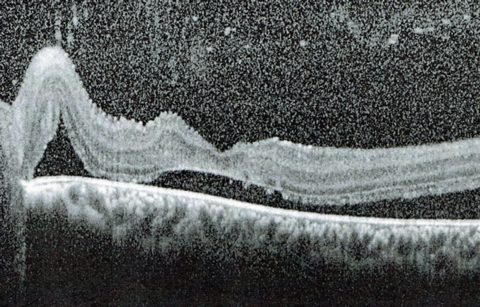 |
| Figs. 5 and 6. These SD-OCT slices show the macula in the right (top) and left eyes. How do you explain these findings? |
Discussion
This disease typically affects young males in their first decade of life. Prevalence of JXRS has been estimated to be anywhere from 1/7000 to 1/2800.2 It is passed on in an X-linked recessive pattern. In other words, only males are affected by the disease while female counterparts are asymptomatic carriers.3 The only known disease gene associated with JXRS to date is Retinoschisin 1 (RS1).2 Generally, entering visual acuities are usually in the range of 20/40 to 20/60 and continue to deteriorate into the third decade of life.2 However, different strands of JXRS exist with varying levels of severity.
Those affected by JXRS will have characteristic cystic changes in the fovea. A stellate pattern of cystic spaces surrounding the foveal area is considered pathognomonic for this condition. This change is secondary to the schisis of the inner retinal region or the nerve fiber layer.2 With time, the cystic spaces begin to coalesce, giving the appearance of foveal atrophy with associated pigmentary changes. Also, peripheral schisis is common in the inferotemporal area. With the coalescence of peripheral schisis areas, vitreous hemorrhages, retinal holes and retinal detachments can occur. These are often secondary to traction in the retinal bands or areas of non-schitic retina.2 If the schisis becomes severe and the vitreous condenses, leukocoria may manifest.1,2 Nonspecific pigmentary changes occur later in the disease as the outer retinal regions become affected by chronic schisis.2
Ancillary testing is extremely helpful and is performed to assist in distinguishing the diagnostic features of JXRS. Scotopic ERG demonstrates an electronegative response with deep negative a-wave and a b-wave that is substantially attenuated and does not return to baseline.1,2 Electronegative ERG has a sensitivity of 100%, as all affected males produce the same results despite age or stage of disease.1 However, there are no reliable or identifying ERG findings in female carriers.1 Electro-oculogram testing is initially normal, but with disease progression affecting the outer retinal layers and RPE, results become abnormal.2 Color vision often reveals proton or tritan deficits.2 Visual field testing can elicit a relative central scotoma, which is related to the foveal schisis, as well as absolute scotomas in regions that correspond to the areas of peripheral retinal schisis (i.e., superonasal scotomas in the presence of inferotemporal schisis).2 Lastly, FA often shows no signs of leakage or staining within the fovea despite apparent cystic changes. In late-stage JXRS, a window defect appears due to the pigment in the fovea area, and capillary nonperfusion in schisis areas of peripheral retina, which may also have associated pigmentary changes.2 As seen in the FA results of our patient, only vascular telangiectatic abnormalities, retinal schisis and retinal detachment were noted; leaking of the vessels was not an issue.
Currently, no treatment exists for either the retinal degeneration or the schisis that occurs from JXRS. However, should a patient experience serious complications from the condition such as a vitreous hemorrhage or retinal detachment, surgical intervention may be warranted.5 As primary care physicians, it is in the best interest of the patient that we best correct vision with polycarbonate lenses and employ the use low vision aids.5
Lately, researchers have studied new treatments for the complications of JXRS. Most notably, one study shows dorzolamide 2% decreases foveal thickness as well as the appearance of the cystic spaces.6 Gene replacement therapy has also been considered as another treatment option. This therapy would aim to replace RS1 and research shows it is successful in knock-out mouse models.5
Regardless, it is ideal for the family of a diagnosed JXRS patient to be genetically tested for RS1 to determine the likelihood of the disease affecting future children and to determine which particular strand of JXRS the patient suffers from, as different strains have varying levels of severity.5
In the case of our patient, he suffered from inferior schisis of the right eye and retinal detachment of the left eye. He underwent a pars plana vitrectomy with scleral buckle in the left eye and experienced no complications. At his most recent visit, the vision in his left eye was best corrected to 20/100. The plan is to proceed with a scleral buckle in the right eye once his left eye is deemed stable. In the meantime, he is to continue using polycarbonate spectacle lenses for correction and protection.
Dr. Diego is a resident at Bascom Palmer in Miami.
1. Spaide R. Diseases of the Retina and Vitreous. London: W.B. Saunders;1999. |
Retina Quiz Answers:
1) b; 2) c; 3) d; 4) a.
