 |
Pheochromocytomas—tumors of the adrenal glands—are a common cause of secondary hypertension. While usually benign, they may also present as, or develop into, a malignancy.1 Because ocular symptoms may be the first or even the only sign, optometrists often see these patients before an internist. Thus, pheochromocytomas must be in the list of differentials whenever a patient presents with ocular evidence of vascular disease.
Pheochromocytoma Basics
The adrenal glands sit on top of each of the two kidneys. Chromaffin cells of the adrenal medulla, or the inner area of gland, are responsible for synthesizing and releasing catecholamines such as dopamine, epinephrine (adrenaline) and norepinephrine (noradrenaline)—the “flight or fight” neurohormones.1
Table 1. Major Causes of Secondary Hypertension1,2,4,5
|
Catecholamines stimulate alpha-adrenergic receptors, resulting in elevated blood pressure (BP), increased cardiac contractility, glycogenolysis, gluconeogenesis and intestinal relaxation.2 They also stimulate beta-adrenergic receptors, causing increased heart rate and contractility. Increased blood pressure secondary to high catecholamine levels can cause headaches, sweating, pounding heart, anxiety and chest pain.2
Pheochromocytomas are tumors of the chromaffin cells. The term pheochromocytoma refers to the color the tumor cells acquire when stained with chromium salts (in Greek, phios means dusky, chroma means color and cytoma means tumor).2 Although pheochromocytomas occur in people of all races, they are diagnosed less frequently in blacks than in whites. They may affect patients of any age, with peak incidence from the third to the fifth decades of life.1 Approximately 10% occur in children.1
The majority of pheochromocytomas are sporadic. Approximately 30% result from inherited mutations, and roughly 10 genes have been identified as sites of mutations.2 Tumors are malignant in 10% of cases, and may be cured completely by surgical removal.1 Pheochromocytomas can occur in combination with other tumors and in some familial syndromes. They have classically been associated with von Hippel-Lindau (VHL) syndrome, multiple endocrine neoplasia type 2 and neurofibromatosis type 1 (NF1).
Approximately 85% of the tumors are located within the adrenal glands. When they occur outside of the adrenal gland, they are termed extra-adrenal pheochromocytomas, or paragangliomas.1,2
Anything that can cause over-activity of the sympathetic nervous system should be on the list of diagnoses to rule out when suspecting a pheochromocytoma.
What to Expect
Patients with pheochromocytoma present with myriad symptoms and signs, giving rise to the tumor’s title of “great masquerader.” In the visual system, hypertensive retinopathy, choroidopathy and optic neuropathy are the main complications.3 In addition, associated conditions such as NF1 and VHL may cause severe neurological manifestations and visual disturbances.
Systemically, the main clinical feature is hypertension, although pheochromocytomas are present in only about 0.2% of those with high blood pressure. Increased blood pressure may be an abrupt, precipitous elevation associated with tachycardia, palpitations, headache, sweating, tremor, postural hypotension, fever, pallor and weight loss. Abdominal or chest pain, nausea and vomiting may occur.1,2,4
Pheochromocytoma-associated hypertensive episodes are caused by increased sympathetic neuronal impulse frequency with excessive release of norepinephrine into the synaptic cleft with each impulse. The secondary hypertension may eventually precipitate congestive heart failure, pulmonary edema, myocardial infarction, ventricular fibrillations and cardiovascular disease.1 Rarely, familial pheochromocytomas may cause no symptoms or signs.4
How to Handle the Situation
A thorough history and meticulous clinical exam are powerful diagnostic tools that can correctly identify the cause of secondary hypertension, and pheochromocytomas in particular (Table 1).1,2,4,5 Although secondary hypertension makes up only about 10% of all hypertensive cases, it is often correctable.1
Hypertension can be confirmed by a simple blood pressure measurement. The laboratory diagnosis of pheochromocytoma is based on the 24-hour urinary excretion/collection of free catecholamines and their metabolites. Plasma metanephrine may also be done. If these are more than two times the normal level, imaging studies are usually necessary to look at the adrenal glands.1,2,4
Abdominal CT scanning has a high accuracy for detecting adrenal masses with a spatial resolution of 1cm or greater.5 MRI is the preferred imaging choice in children and pregnant/lactating women, with a sensitivity of up to 100% in detecting adrenal pheochromocytoma.5
Scintigraphy is reserved for biochemically confirmed cases in which CT or MRI do not show a tumor.5
Positron emission tomography (PET) scanning is another useful technique.5 Because cancer cells often take up more glucose than normal cells, PET imaging is particularly useful for visualizing them.
Surgical resection is the treatment of choice and usually cures the hypertension and ocular side effects. Preoperative treatment with alpha and beta blockers is required to control blood pressure and prevent intraoperative hypertensive crises. Until the tumor is removed, blood pressure control is a top priority.4
Both malignant and benign pheochromocytomas can recur after surgery. The statistics vary between studies, but recurrence rates average around 10%.2,4 Therefore, long-term follow-up care after surgery is essential. In the low percent of these already rare tumors in which malignant behavior is evident, survival may still be quite prolonged, as the pace of the disease is often slow. Participation in clinical trials of new therapies is strongly encouraged in the unfortunate case of metastatic pheochromocytoma.5 This will direct the plan of management, thus preventing treatment delay and potentially serious complications.
Whenever the diagnosis is in doubt, biochemical testing can establish the presence or absence of a pheochromocytoma, and localization with neuroimaging is almost always possible. While an uncommon finding, ODs must keep pheochromocytomas on their list of differentials, especially for patients who present with sustained or paroxysmal hypertension and any manifestations suggesting excess catecholamines. Quick diagnosis and proper comanagement are key to reduce the negative effects of secondary hypertension.
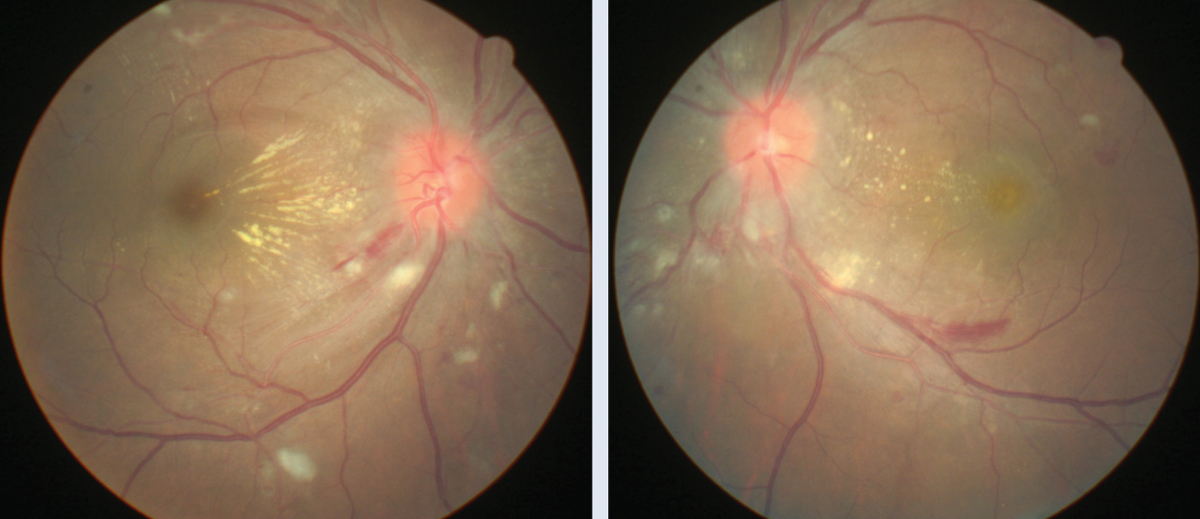
Case ExampleA 30-year-old female presented with complaints of moderate bilateral visual blur associated with headaches and photophobia. She stated that she had been to the emergency room one week prior to address the onset of heart palpitations. She was treated for severe hypertension and released for further medical management by her primary care provider (PCP). During her visit, her pinhole visual acuity was 20/40 OD and 20/50 OS. Ophthalmoscopy showed bilateral optic disc edema, soft exudates, macular star, flame-shaped hemorrhages and arterial narrowing (Figures 1 and 2). Her blood pressure measured 141/92mm Hg. An ocular diagnosis of Grade 4 hypertensive retinopathy was established, and the patient was referred back to her PCP for continued treatment. Due to the patient’s young age, we needed to rule out secondary hypertension. Neuroimaging studies revealed a right adrenal gland mass. After ablation, the tumor proved to be a pheochromocytoma, and immunohistochemistry showed dopamine secretion. In addition to continued medical treatment, the patient will return to our clinic in one month to assess visual function and monitor ocular health. |
1. Waguespack SG, Rich T, Grubbs E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010;95(5):2023-37. |
