In the United States, approximately 4,200 persons are affected by a corneal dystrophy, according to research from Prevent Blindness, the National Institute of Special Education and the National Science Foundation.
The corneal dystrophies are bilateral, usually symmetric inherited diseases of the cornea. They result in changes that include opacities of various shapes and sizes that affect all corneal layers. These opacities typically affect the central cornea early in life and increase and spread to the periphery. Corneal dystrophies affect different populations to varying degrees. For example, 4% of patients in Saudi Arabia and 12.6% of patients in Japan who underwent corneal transplants had corneal dystrophies.1,2
Corneal dystrophies do not have marked signs of inflammation, such as neovascularization, and most are not associated with systemic disease. Most dystrophies are inherited as autosomal-dominant traits (50% of family members are expected to be affected), but a few corneal dystrophies are inherited as autosomal- recessive traits and have more se-vere outcomes. Also, some dystrophies are sex-linked.
Currently, the classification of the corneal dystrophies is based on the affected layer of the cornea; this classification, however, does not re-flect the genomics of each dystrophy. Some corneal dystrophies with differing phenotypic presentations that affect different corneal layers are caused by different mutations in the same gene. This fact may change the way we classify corneal dystrophies in the future. Also, the almost daily discovery of new mutations may someday lead to gene therapy and/or non-surgical means of treating these dystrophies.
In this third and final part of our Genetic Disease series, we will discuss the clinical presentations, treatments and genetic mutations of the most common corneal dystrophies.
Epithelial Dystrophies
There are two common epithelial dystrophies:
Epithelial basement membrane corneal dystrophy (EBMD). This dystrophy, also known as Cogans microcystic dystrophy or map-dot-fingerprint (MDF) dystrophy, is likely the most common anterior basement membrane dystrophy.3 As its name implies, it is characterized by three types of changes: map-like, dot-like and swirls (fingerprints). It is not a true dystrophy in the sense that similar changes are also seen in 76% of the normal population older than 50 years.4 Pedigrees with autosomal-dominant inheritance have been reported.

Microcysts, fingerprint swirls and map-like changes characterize epithelial basement membrane dystrophy.
These changes are caused by a build-up of abnormal basement membrane material. Patients who have EBMD are prone to recurrent corneal erosions (RCE) because their epithelium is unstable due to the presence of a thickened basement membrane. Other symptoms of this epithelial dystrophy: photophobia, blurred vision, pain and contact lens intolerance.
Treatment of RCEs includes hy-pertonic solutions and ointments, patching, mechanical debridement of loosened epithelium, bandage contact lenses, and phototherapeutic keratectomy (PTK) when there is significant visual compromise from irregular astigmatism.
Mutations that cause EBMD have been localized to the transforming growth factor, beta-induced (TGFBI or BIGH3) gene on the long arm of chromosome 5 (5q31).5 This gene, first mapped to chromosome 5 in 1994 and identified in 1997, encodes for an adhesion protein known as keratoepithelin, which is secreted by the corneal epithelium and found in the stroma.6,7 Mutations in this gene result in the formation of corneal opacities that contain keratoepithelin. Mutations in TGFBI are also associated with other corneal dystrophies that affect the Bowmans layer and stroma as well as the corneal epithelium.
Meesmans dystrophy. This autosomal-dominant epithelial dystrophy results in the formation of diffuse intraepithelial vesicles or microcysts. These clear cysts present in the first decade of life and contain keratin. Patients are usually asymptomatic until the fourth or fifth decade, when they may develop irregular astigmatism and blurred vision. If the cysts rupture, patients will have symptoms of photophobia and tearing. There is some evidence that soft contact lenses may decrease the number of microcysts.8
Mutations in two specific keratin genes, KRT3 on chromosome 12 and KRT12 on chromosome 17, have been associated with Mees-mans dystrophy.9 Mutations in these genes, which are responsible for the structural integrity of the epithelium, lead to the abnormal assembly of the filaments and result in an unstable epithelium that cannot withstand even mild trauma to the cornea.
Bowmans Layer Dystrophies
Reis-Bucklers corneal dystrophy (corneal dystrophy of Bowmans layer or CDB1) presents in the first decade of life with central gray-white opacifications that are often ring-shaped. These eventually spread across the corneal surface, giving it a ground-glass appearance.
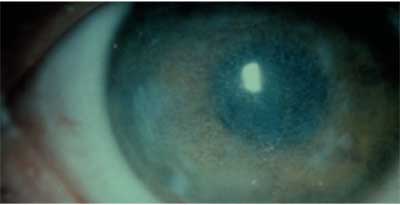
2. Ring-shaped opacities in advanced Reis-Buckler dystrophy give the corneal surface a ground-glass appearance.
In this dystrophy, the basement membrane and Bowmans layer are replaced by masses of curly filaments. The epithelium is irregular and thin, and patients are subject to RCEs. Although RCE attacks lessen with increasing age, vision loss can be progressive due to increasing corneal opacification over time.
Treatments for Reis-Bucklers corneal dystrophy include epithelial debridement or PTK, although lamellar or penetrating keratoplasty may be required in severe cases in which there is a lot of opacification of the cornea; recurrences of the dystrophic changes can occur in the grafts. Mutations that cause this dystrophy have also been localized to the TGFB1 gene.
There are two other dystrophies that affect Bowmans layer: Thiel-Behnke dystrophy (affecting only Bowmans layer) and Grayson-Wilbrandt dystrophy (a later-onset dystrophy associated with few RCEs). These dystrophies were previously thought to be distinct and separate conditions but are now considered to be variants of Reis-Bucklers dystrophy.10
Stromal Dystrophies
The stroma constitutes 90% of the corneal thickness, and there are more stromal corneal dystrophies than dystrophies affecting any other layer of the cornea. There are genotypic similarities between some stromal dystrophies and those that affect the epithelial and Bowmans layers, even though there are phenotypic differences. The more commonly encountered stromal dystrophies are:
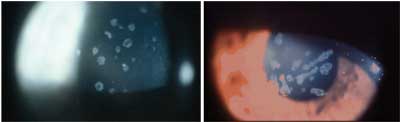 | |
| Granular dystrophy in a 20-year-old woman (left) and in the womans mother, presents with crumb-like, ring-shaped powdery opacities. | |
Most patients do not require treatment, except for the RCEs that can occur in this dystrophy. In advanced stages in which the stroma between the granular opacities is no longer clear and/or the granular lesions have coalesced to form linear opacities that compromise vision, PTK, lamellar keratoplasty and sometimes penetrating keratoplasty (PK) are necessary. Granular opacities are possible, but they start in the periphery of the graft.
Several mutations on the TGFB1 gene on chromosome 5q31 are associated with granular dystrophythe same gene associated with the previously discussed dystrophies.
Avellino dystrophy. This type of granular dystrophy, discovered in 1988 in families of Italian de-scent (from Avellino, Italy), presents with both granular and lattice-type opacities. The granular opacities, which are composed of hyaline, appear earlier, and the lattice-type lines composed of amyloid appear later.11 These patients usually have more RCEs than patients who have granular dystrophy.12
One mutation (R124H) on the same TGFB1 gene is responsible for causing this dystrophy.7
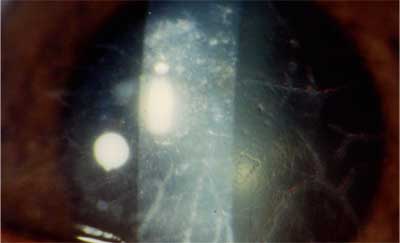 |
| All types of lattice dystrophy are characterized by the appearance of branching lattice lines made up of amyloid material deposits in the stroma. |
Type II lattice dystrophy is one of the few corneal dystrophies associated with a systemic disease: familial amyloidosis. Type II lattice dystrophy is actually a manifestation of a systemic disease (called Meretoja syndrome).13 This disease is associated with amyloid deposition in a few tissues, such as the peripheral nerves (causing palsies), skin (causing dryness and itchiness), the face (causing a mask-like appearance) the brain and the cornea. There are fewer lattice lines in Type II than in Type I. They appear later (after age 20) and are more peripheral than central.
The mutation for this stromal dystrophy occurs in the gelsolin gene (GSN), on chromosome 9 (9q34). This is clearly a different type of lattice dystrophy because it is a later-onset dystrophy, the result of amyloid deposition in the body, and lines are thinner and more peripheral.14 This gene encodes for a filament modulation protein.
There are other types of lattice dystrophies that have subsequently been described in the literature based on mode of inheritance (Type IIIb is autosomal recessive) and delayed onset (Type IIIa); additional types have been described in various pedigrees. Type IIIa has also been linked to a mutation in the TGFB1 gene.
Treatment for lattice dystrophy is based on the severity of the disease and the patients symptoms. RCEs are associated with many types of lattice dystrophy. If visual acuity is compromised, PK is then required, although recurrence of the RCEs is still possible.
Macular dystrophy. This is one of the most visually debilitating of the dystrophies and is inherited as an autosomal-recessive trait.15 Milky white stromal opacities made of glycosaminoglycan proteins appear early and spread quickly (figure 6). Visual acuity is compromised by age 40, and PK is required.15 However, recurrences in the donor graft are possible.16
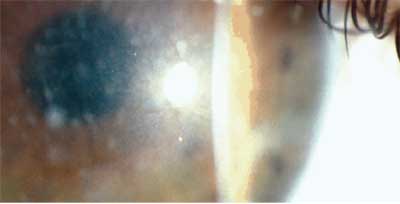 |
| Milky white stromal opacities made of glycosaminoglycan appear early and spread quickly in macular corneal dystrophy. |
There are two types of macular dystrophy based on the synthesis of keratin sulfate (KS). This protein is necessary for the production of corneal proteoglycans. In Type I macular dystrophy, KS is not synthesized. In Type II, KS is synthesized but is below normal levels.
Types I and II have been linked to the CHST6 gene on the long arm of chromosome 16 (16q22).17 This gene codes for an enzyme needed in the sulfation of keratin. Many mutations on this gene that cause macular dystrophy have been identified.17
Posterior Corneal Dystrophies
There are three types of posterior (Descemets membrane and endothelium) corneal dystrophies:
Fuchs endothelial dystrophy. This is a later-onset dystrophy that affects women more than men, although there is an early-onset (between 20 and 40 years of age) hereditary form called early-onset Fuchs dystrophy.18 Early clinical features include corneal guttata, which are localized excrescences, or warts, in Descemets membrane that lead to endothelial dysfunction. This process causes stromal and epithelial edema and eventually corneal fibrosis. The corneal edema is usually worse in the morning, as closed eyes during sleep do not allow for evaporation of the excess fluid from the cornea.
Treatment consists of hyperosmotic agents, usually q.i.d., during the day and ointment at night. Also, a hair dryer can help evaporate excess fluid from the cornea. As fibrotic scarring increases, surgical intervention may be required. PK once was the only treatment of choice for patients at this stage. Now, other less invasive treatments, such as deep lamellar endothelial keratoplasty and Descemets stripping endothelial keratoplasty are being attempted. Fuchs dystrophy (or any other endothelial dystrophy) must be ruled out in patients who are considering cataract extraction because the cataract surgery can cause a cornea with Fuchs dystrophy to decompensate, leading to a resulting bullous keratopathy and poor vision.
Although many cases of Fuchs dystrophy occur without family history, there are reports of autosomal-dominant inheritance. Mutations in the COL8A2 gene on the short arm of chromosome 1 (1p34.3-p32) have been associated with an early-onset (third to fifth decade) type of Fuchs dystrophy whereas the other type of Fuchs affects individuals in the fifth to seventh decades.19 This gene encodes for collagen, which may play a structural role in Descemets membrane.20 A later-onset type of Fuchs dystrophy has been linked to a new genetic locus on chromosome 13.21
Posterior polymorphous dystrophy (PPD). PPD is an autosomal dominant dystrophy that starts earlier than Fuchs dystrophy and follows a more benign course.14 It is characterized by the early appearance of vesicle-like lesions, bands or diffuse opacities. These opacities represent more diffuse thickenings in Descemets membranea contrast to the localized thickenings seen in Fuchs dystrophy. Because of the larger areas affected, PPD can also result in adhesions between the iris and the cornea (peripheral anterior synechiae). These patients must be monitored for the development of glaucoma because many or wide areas of peripheral anterior synechiae can compromise the trabecular meshwork and angle. Corneal edema is also a feature of PPD.
Management of PPD is the same as it is for Fuchs dystrophy.
PPD has been mapped to chromosome 20 (20p11.2-q11.2), and three mutations have been identified in the VSX1 gene in the same locus.22,23 Mutations in this gene have also been associated with keratoconus.24
Congenital hereditary endothelial dystrophy (CHED). This rare endothelial dystrophy causes corneal edema from birth or during infancy. There are two types: Type I (CHED 1) is inherited as an autosomal-dominant trait that presents with clear corneas at birth. Type II (CHED II) is more common but more severe. It is inherited as an autosomal-recessive trait, associated with nystagmus and corneal edema from birth.
Both forms map to the short arm of chromosome 20, along with PPD.25 Because these corneas are cloudy early on (at birth for CHED II and during infancy in CHED I), PK is the only treatment to prevent deprivation amblyopia.
Your Role
Once you diagnose a corneal dystrophy:
Detail what patients can ex-pect regarding progression and treatment.
Notify patients that other family members should undergo an examination.
Consider genetic testing.
Genetic testing is useful to patients and doctors because: It provides clinicians with the information they need to correctly diagnose a dystrophy. In many cases, the identification of a specific mutation will provide more accurate information about the course or progression of the condition and the patients response to surgical intervention. It also provides information that facilitates genetic counseling if it is sought. Finally, genetic testing helps to identify patients for clinical trials. Studies of inherited diseases are more likely to succeed when the mutations have been accurately identified in the enrolled subjects.
Most corneal dystrophies are diagnosed based on the clinical picture, detailed family history, examination of family members and construction of a pedigree. However, if genetic testing is desired, genetic testing is only available currently for mutations in one genethe TGFB1 gene. Testing of mutations on this gene can be offered if the clinician is unsure of the diagnosis or if the family requests it. The cost is about $125 with a turnaround time of eight to 10 weeks.
The future of the molecular genetics of corneal dystrophies lies not only in identification of the dystrophy but in the possibility of therapeutic intervention with drugs or gene therapy to prevent or delay the need for more invasive surgical treatment.
Dr. Bass is a distinguished teaching professor at the State University of New York State College of Optometry. She teaches the posterior segment disease course in the professional program, and she teaches in the glaucoma, retina and cornea clinic.
|
Genetics For Some of The Corneal Dystrophies | |||
| Dystrophy | Description | Affected Gene/Chromosome | Treatment |
| Epithelial Dystrophies | |||
|
Epithelial basement membrane dystrophy |
Map-like, fingerprint swirls; dot-like microcysts | TGFB1/5 in hereditary cases | Treat recurrent corneal erosions (RCEs) |
| Meesmans dystrophy | Diffuse clear microcysts | KRT3/12q13 and KRT12/17q12 | Corneal lubrication |
|
Bowmans Dystrophies | |||
| Reis-Buckler dystrophy | ring-shaped opacities | TGBF1/5q31 | Treat RCEs; may require phototherapeutic keratectomy (PTK) or debridement |
| Stromal Dystrophies | |||
| Granular dystrophy | Central crumb-shaped or ring-shaped opacities made of hyaline | TGBF1/5q31 | PTK or penetrating keratoplasty (PK) in advanced cases |
| Lattice dystrophy Type I | Thin central lattice lines made of amyloid | TGBF1/5q31 | PK in advanced cases |
| Lattice dystrophy Type II | Thicker lattice lines start peripherally; associated with systemic familial amyloidosis | GSN/9q34 | PK in advanced cases |
| Avellino dystrophy | Granular changes followed by lattice lines later on | TGBF1/5q31 | PTK or PK in advanced cases |
| Macular dystrophy | Milky white lesions composed of glycosaminoglycan; autosomal-recessive inheritance with more severe vision loss | CHST6/16q22 | PK by age 40 |
| Endothelial Dystrophies | |||
| Fuchs dystrophy | Guttata (localized thickenings in Descemets membrane) followed by stromal and epithelial edema and corneal fibrosis | COL8A2/1p34.3-p32) in early onset and a late-onset Fuchs on locus 13pTel-13q12.13 | Hyperosmotic agents, hair dryerfor edema; eventual PK or endothelial keratoplasty |
| Posterior polymorphous dystrophy | Vesicles, bands of thickened Descemets membrane, corneal edema, and peripheral anterior synechiae | VSX1/20p11.2-q11.2 | Similar to Fuchs; watch IOP |
|
Congenital hereditary endothelial dystrophy |
Corneal edema at birth or in infancy | Short arm of chromosome 20 along with PPD | PK early on |
1. al Faran MF, Tabbara KF. Corneal dystrophies among pa-tients undergoing keratoplasty in Saudi Arabia. Cornea 1991 Jan;10(1):13-6
2. Santo RM, Yazmaguchi T, Kanai, et al. Clinical and histo-pathological features of corneal dystrophies in Japan. Opthalmology 1995 Apr;102(4):557-67.
3. Smolin G. Corneal dystrophies and degenerations. In: Smolin G, Thoft RA (eds). The Cornea. 3rd Ed Boston: Little, Brown and Co.,1994: 509.
4. Werblin TP, Hirst LW, Stark WJ, Maumenee IH. Prevalence of map-dot-fingerprint change in the cornea. Br J Ophthalmol 1980 Jun;65:401.
5. Boutboul S, Black GC, Moore JE, et al. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFB1/BIGH3. Hum Mutat 2006 Jun;27(6):553-7.
6. Stone EM, Mathers WD, Rosenwasser GO, et. al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet 1994 Jan;6(1):47-51.
7. Munier FL, Korvatska E, Djemai A, et al. Keratoepithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 1997 Mar;15(3):247-51.
8. Bourne WM. Soft contact lens wear decreases epithelial microcysts in Meesmanns corneal dystrophy. Trans Am Ophthalmol Soc 1986;84:170.
9. Irvine AD, Corden LD, Swensson O, et.al. Mutations in cor-nea-specific keratin K3 or K12 genes cause Meesmans corneal dystrophy. Nat Genet 1997 Jun;16(2):184-7.
10. Chang SW, Tuli S., Azar DT. Corneal dystrophies. In: Traboulsi EI (ed). Genetic Diseases of the Eye. New York: Oxford University Press; 1998:223
11. Folberg R, Alfonso E, Croxatto JO, et al. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits. A study of these families. Ophthalmology 1988 Jan;95(1): 46-51.
12. De Sousa LB, Mannus MJ. The stromal dystrophies. In: Krachmer JH, Mannis MJ et al(eds) Cornea, 2nd ed. Philadelphia: Elsevier Mosby; 2005:916).
13. Pieramici SF, Afshari NA. Genetics of corneal dystrophies: the evolving landscape. Curr Opin Ophthalmol 2006 Aug;17 (4):361-6.
14. Klintworth GK. The molecular genetics of the corneal dystrophies-current status. Front Biosci 2003 May 1;8:687-713.
15. Smolin G. Corneal dystrophies and degenerations. In: Smolin G, Thoft RA (eds). The Cornea. 3rd ed. Boston: Little, Brown and Co.; 1994: 514
16. Klintworth GK, Reed J, Stainer GA, Binder PS. Recurrence of macular corneal dystrophy within grafts. Am J Ophthalmol 1983 Jan; 95(1):60-72.
17. Klintworth, GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular genetic review. Mol Vis 2006 Mar 10; 12:159-176.
18. Gottsch JD, Sundin OH, Liu SH, et. al. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of Fuchs corneal dystrophy. Invest Ophthal Vis Sci 2005; 46: 1934-9.
19. Biswas S, Munier FL, Yardley J, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 2001 Oct 1;10(21):2415-23.
20. Levy SG, Moss J, et. Al. The composition of wide-spaced collagen in normal and diseased Descemets membrane. Curr Eye Res 1996;15:45-52.
21. Sundin OH, Jun AS, Broman KW, et.al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest Ophthalmol Vis Sci 2006 Jan;47(1):140-5.
22. Heon E, Mathers WD, Alward WL, et al. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet 1995 Mar; 4(3):485-8.
23. Heon E, Greenberg A, Kopp KK, et al. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 2002 May 1;11(9):1029-36.
24. Busceglia L, Ciashetti M. VSX1 mutational analysis in a series of Italian patients affected by keratoconus: detection of a novel mutation. Invest Ophthalmol Vis Sci 2005 46(1):39-45.
25. Toma NM, Ebenezer ND, Inglehearn CF, et. al. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet 1995 Dec;4(12):2395-8.
