She reported trouble seeing clearly at distance, especially when attempting to read road signs while driving at night. Further, she said that her twin sister and older brother also have eye problems. Her medical history was unremarkable.
On examination, her best-corrected visual acuity measured 20/60 OD and 20/40 OS. Confrontation visual fields were full to finger counting OU. Her pupils were equally round and reactive to light, with no evidence of afferent defect. Extraocular motility testing was normal in both eyes. Her anterior segment examination was unremarkable OU.
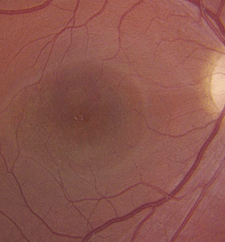
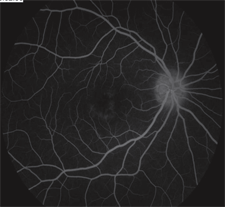
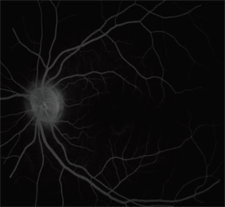
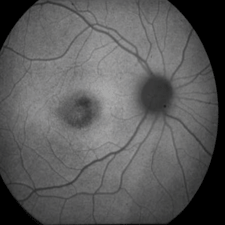
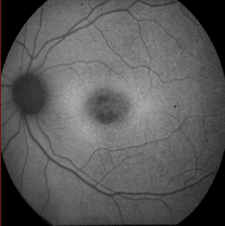
The dilated fundus exam revealed small cups with good rim coloration and perfusion OU. She exhibited mild retinal pigment epithelium (RPE) mottling in both maculae. Foveal light reflex was visible OU. The vessels and peripheral retinae were normal in both eyes. The posterior pole can be viewed in figures 1 and 2. Additionally, we performed fluorescein angiography (figures 3 and 4) and fundus autofluoresence (figures 5 and 6).
|
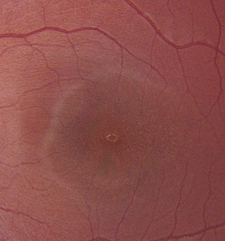 |
| 1, 2. Fundus photographs of our patient's posterior poles (OD left, OS right).
| |
 |
 |
|
3, 4. What does the fluorescein angiogram confirm (OD left, OS right)?
| |
 |
 |
| 5, 6. Fundus autofluoresence of our patient (OD left, OS right). What does this reveal?
| |
Take the Retina Quiz
1. How would you describe the significant findings seen in the fluorescein angiogram?a. Unremarkable.
b. Marked staining and hyperfluorescence.
c. Blockage of the background choroidal fluorescence.
d. Hyperfluorescence of the macula in a “bull’s-eye” pattern.
2. What is the correct diagnosis?
a. Occult retinal dystrophy.
b. Stargardt disease.
c. Cone dystrophy.
d. Malingering.
3. Which gene has been linked to this condition?
a. Complement factor H (CFH).
b. Rhodopsin gene (RHO).
c. ARMS2.
d. ABCA4.
4. What is our patient’s prognosis?
a. Her vision should improve.
b. Her vision will likely remain stable without further loss.
c. Slow, progressive vision loss that will stabilize near 20/200.
d. Slow, progressive vision loss that will lead to blindness.
Discussion
When you look at our patient’s retinae, they appear fairly normal. There is mild RPE mottling in the maculae––but, beyond that, they seem healthy. Additionally, her posterior poles and peripheral retinae look pretty good. So, how do you explain the reduced acuity? In this case, our patient’s family history provides the clues.Remember––her twin sister also experienced significant visual acuity reduction. A further review of her sister’s ocular history revealed the presence of bilateral, central atrophic scars that exhibited a “beaten metal” appearance. Fortunately, our patient’s maculae do not appear nearly as damaged as those of her twin. Nevertheless, we must keep in mind that such a beaten metal appearance is highly indicative of a visually devastating genetic condition––Stargardt disease.
Stargardt disease is the most common hereditary macular dystrophy.1,2 It is an autosomal recessive condition that presents in approximately one in 10,000 individuals worldwide.1 Generally, it manifests during patients’ teenage years and progresses throughout their early 20s.
However, there have been reports of the condition developing in older adults as well.1,2 Stargardt disease is linked to a mutation in the ABCA4 gene, which codes an energy-dependent transmembrane protein localized in the discs of the rod and cone outer segments.2 It is considered to be a lipofuscin storage disease. Excess lipofuscin in the RPE makes the retina appear deep red or vermillion, as well as obscures some choroidal detail on clinical examination. Indeed, if you look carefully at the coloration of our patient’s retinae, they appear to have a deep red appearance
Further, the choroidal vasculature that often can be seen through the RPE is absent. Given this information alone, however, it would be difficult to make a diagnosis. The diagnosis of Stargardt disease is usually made based on both the clinical appearance and an index of suspicion. The difficulty in diagnosing the condition solely based upon clinical appearance is that it varies from patient to patient. The maculae can appear normal (as was the case in our patient), or they can exhibit atrophic RPE changes that are often described as beaten metal or “beaten bronze.”
Further, patients may have macular atrophy with or without scattered, yellow-white flecks in the posterior pole and periphery. Alternatively, they may present with flecks in the absence of macular atrophy. Fluorescein angiography is considered the gold standard diagnostic test for Stargardt disease. Because of the increased levels of lipofuscin, the staining pattern will reveal blockage of the background fluorescence, resulting in a quiet choroidal appearance. This is the case in our patient. More recently, fundus autofluorescence has emerged as a useful tool for evaluating the extent of Stargardt disease. This diagnostic technology enables the visualization of A2E, a critical component of lipofuscin.3
Within our patient’s maculae, we saw a circular area of hypofluorescence that may indicate early RPE atrophy and photoreceptor cell loss. In the relatively near future, our patient likely will develop visible atrophy, like her sister.
There is no treatment for Stargardt disease, beyond ensuring that the patient’s optical needs are met. At present, our patient’s visual acuity is still pretty good. This will not last very long, however, because the disease likely will continue to progress.
Quiz Answers: 1) c; 2) b; 3) d; 4) c
1. Blacharski PA. Fundus Flavimaculatus. Retinal Dystrophies and Degenerations. New York: Raven Press; 1988:135-59.2. Deutman AF, Hoyng CB, van Lith-Verhoeven JJC. Macuar dystrophies. In: Ryan SJ (ed.). Retina. Vol. II, 4th ed. St. Louis: Mosby; 2006:1160-209.3. Gomes NL, Greenstein VC, Carlson JN, et al. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009 Aug;50(8):3953-9.
