Annual Retina ReportCheck out the other feature articles in this month's report:Reconsider Your Approach to Diabetic Retinopathy Macula Exam Tips and Tricks Setting Patient Expectations for Anti-VEGF Therapy Sizing Up Geographic Atrophy |
Retinitis pigmentosa (RP) is a group of inherited diseases involving progressive retinal degeneration of retinal pigmented epithelial cells and photoreceptors. The disease affects approximately one in 3,000 to 5,000 people without sex predilection and requires frequent examination.1 Inheritance patterns of RP are most often autosomal recessive (AR), autosomal dominant (AD) and X-linked.
Because there is variability between the genotype and its phenotypic expression, the prognosis cannot be determined by the inheritance pattern alone. However, there is a general correlation between age-related visual acuity and mode of inheritance.2 In cases where only one person in a family is affected, the disorder is described as simplex, which may occur through a new gene mutation. As it stands, researchers believe approximately 60 genes cause RP with variable inheritance patterns.3 Primary RP is the predominant form of the disease restricted to ocular manifestations. Secondary RP occurs as a manifestation of systemic syndromes, most notably Usher syndrome.
Presentation
Clinical manifestations of RP are generally characterized by bilateral nyctalopia, constricted visual fields, pigment atrophy, pigment clumping in a bone-spicule configuration and rod-cone dysfunction.4 Symptoms of decreased peripheral vision and nyctalopia typically begin in the first and second decades of life and steadily worsen.4
Nevertheless, genotypic variability often gives rise to variable phenotypic observations. As such, two individuals with AR-RP may produce variable expression patterns.4 Less common forms of the disease include unilateral RP and RP sine pigmento, the latter of which is characterized by a normal-appearing fundus with an abnormal electroretinogram (ERG).
Early funduscopic signs of RP include mild arteriole attenuation, pigment dusting in the vitreous, and retinal pigment dispersion of the anterior and mid-peripheral retina.5 At this stage, an ERG may show flattening of both a-waves (photoreceptor) and b-waves (inner retinal cells).5 Generally, formal visual field testing will display a mid-peripheral scotoma. Early fundus autofluorescence (FAF) may demonstrate both hyper- and hypofluorescence of the anterior and mid-periphery signaling retinal pigment epithelial (RPE) distress. With time, melanin will migrate anteriorly within the intraretinal space in a bone-spicula configuration.
Patients with advanced RP typically exhibit marked vessel attenuation, severely constricted visual fields, optic atrophy, posterior subcapsular cataracts, cystoid macular edema (CME), central vision loss, dyschromatopsia and photophobia.5 In advanced-stage RP, FAF will usually display a hyperfluorescent parafoveal ring, signaling preservation of the ellipsoid zone (EZ) as visualized on OCT.6
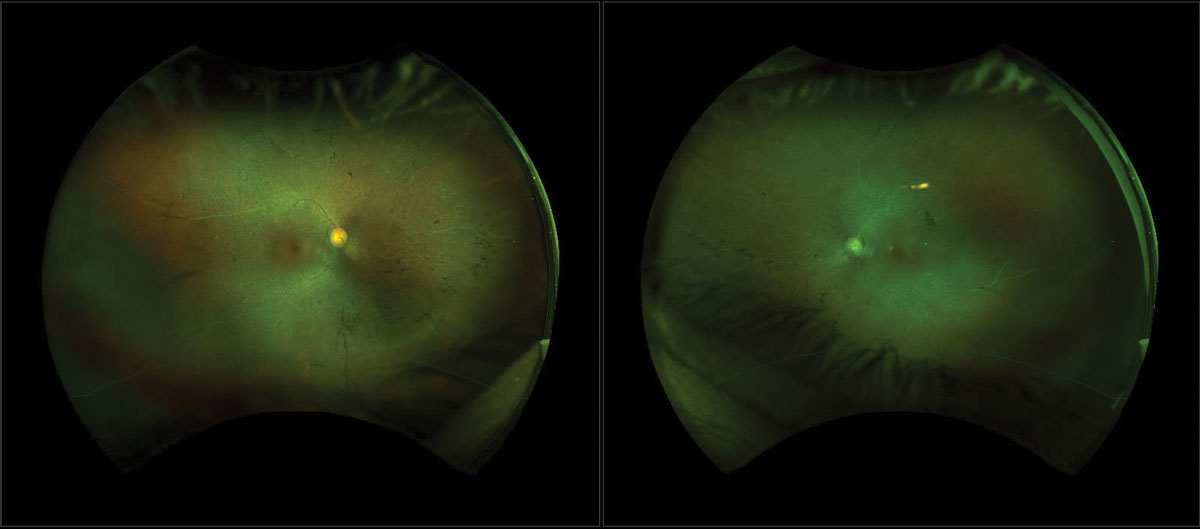 |
| Fig. 1. FAF that displays a hyperfluorescent ring and hypopigmentation in the mid-periphery can be related to late-stage RP. Click image to enlarge. |
Examination
The American Academy of Ophthalmology recommends that the clinical assessment of suspected inherited retinal dystrophies include a comprehensive examination along with imaging (color fundus photos, fundus autofluorescence and SD-OCT), formal visual fields, full field ERG and molecular genetic testing.7
A clinical exam should begin with a thorough evaluation of the patient’s ocular, medical, social and family histories, including identifying any past history of retinotoxic medications that may help differentiate RP from alternative etiologies.
Annual comprehensive exams are required to identify progression as well as potential secondary complications, such as posterior subcapsular cataracts, optic atrophy, optic disc drusen and CME.7 We prefer the widefield FAF over the standard color photograph, as it can demonstrate photoreceptor degeneration (hypofluorescence), as well as accumulation of lipofuscin (hyperfluorescence). In addition, a large number of patients will exhibit a hyperfluorescent Robson-Holder ring. The diameter of the ring on FAF corresponds well with retinal sensitivity and constriction, as demonstrated by formal visual field testing.8 While the ERG remains the standard of care for diagnosis, FAF can be used in lieu of ERG to monitor progression, especially in later stages of the disease when ERG is less reliable.9
As the disease progresses, OCT is sensitive at identifying early changes in the macula responsible for central vision loss. Close inspection of the EZ can help identify early structural abnormalities.10 Discontinuity of the EZ is followed by shortening of the ELM and RPE.11 To quantify the extent and degree of peripheral vision, perform visual field testing (HVF 30-2).
Early changes usually begin with isolated, mid-peripheral scotomas that gradually coalesce to form a partial ring scotoma. As the disease progresses, the outer edge of the ring expands to the periphery, while the inner edge constricts centrally. Examine patients who lack central fixation with the full-field stimulus test.7
ERG measures the electrical potential of the photoreceptors and can establish the diagnosis. In early stages of the disease, the amplitudes of the a- and b-waves are diminished. With progression, the amplitudes may become extinguished. At this stage, multifocal ERG and microperimetry are effective at objectively measuring functional vision.12,13
Treatments
Since RP has no cure, most therapies are limited. Studies show that vitamin A, lutein and docosahexaenoic acid (DHA) supplements may reduce the risk of progression.14-19 Counsel patients with primary RP and those with Usher syndrome on the potential therapeutic benefits of daily intake of vitamin A palmitate 15,000 IU, lutein (12mg), two ounces of omega-3 rich foods or a DHA supplement (200mg), and to avoid supplemental vitamin E as it hastens the progression of the disease.18-20
Prior to initiating therapy, we recommend that patients undergo medical evaluation to assess fasting serum vitamin A, red blood cell DHA levels and liver enzymes. Following a normal baseline, perform blood tests annually. Aside from supplementation, a number of studies are exploring new treatment options. Novel therapies are aimed at preserving and restoring vision in patients with RP. Some of these methods include gene therapy, autoserum, stem cell therapy and visual prostheses.21-23 Most notably, Luxturna (voretigene neparvovec, Spark Therapeutics) has received FDA approval for treating patients with biallelic RPE65-mediated retinal dystrophy.24
Low vision aids can improve visual performance in patients with RP. Traditional optical aids include telescopes, reverse telescopes, wide-field and high-intensity flashlights, CPF 550 lenses (Corning), hand-held magnifiers, stand magnifiers, half-eye base-in prism lenses and electronic devices. Additionally, the Argus II retinal prosthesis has received FDA-approval for a limited number of patients with vision of light perception or worse.25
Case Presentation
A 59-year-old Hispanic female presented with progressive vision loss and night blindness since childhood. She was first seen 10 years ago in Puerto Rico for a gradual decrease in vision and difficulty driving at night attributed to cataracts. Shortly after cataract surgery, she was lost to follow-up,with no records to review. During this period, the patient began to notice a gradual decline of peripheral vision in both eyes and significant loss of central vision in the left eye. Her ocular history was positive for cataract surgery, mild hyperopia and presbyopia OU. She had no history of ocular inflammation or eye trauma. Her medical history was positive for hypothyroidism, which she treated with levothyroxine. A complete review of systems was unremarkable for inflammatory disease, prior infection, infantile illness or sexually transmitted disease. Her family and social histories were unremarkable.
Best-corrected visual acuity was 20/50 OD and light perception OS. Pupils were round and slowly reactive to light with a relative afferent defect OS. Confrontation visual fields were severely constricted OU. Color vision, measured with Ishihara plates, was reduced to 8/10 OD and 0/10 OS. The posterior vitreous was detached OU. The estimated cup-to-disc ratio was 0.50 OD and 0.70 OS. The color of the disc was normal OD and diffusely pale OS. A dilated fundus exam revealed diffuse atrophy and clumping of the RPE OU. Examination of the macula revealed cystic elevation and granular pigmentary changes in both eyes. Retinal arterioles were moderately attenuated OD and severely attenuated OS. Retinal photography was performed to document the findings.
Fundus autofluorescence revealed a parafoveal hyperfluorescent ring and hypopigmentation in the mid-periphery (Figure 1). SD-OCT demonstrated CME as well as significant disruption of the EZ, external limiting membrane (ELM) and RPE (Figure 2). Automated Humphrey 30-2 standard visual field showed severe defects in all four quadrants OU with sparing of central fixation OD.
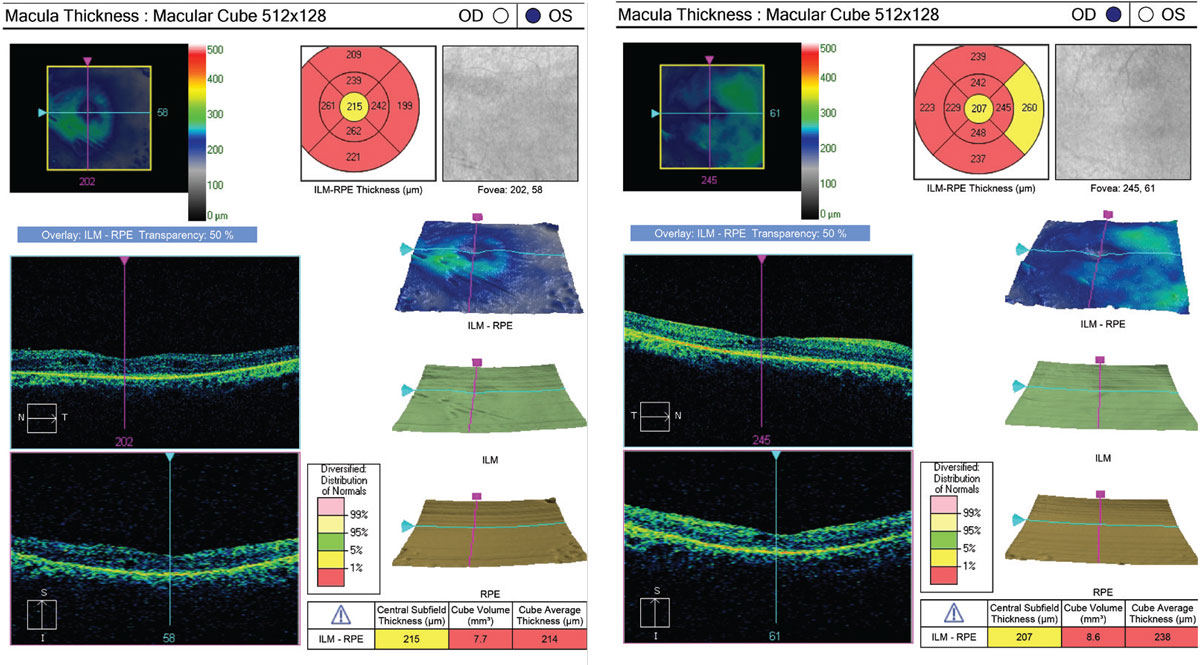 |
| Fig. 2. OCT is sensitive at identifying CME responsible for central vision loss. Close inspection of the EZ can also help identify structural abnormalities suggestive of RP. Click image to enlarge. |
Based on the patient’s history and clinical presentation, we established a tentative RP diagnosis OU with secondary optic nerve cupping and pallor OS. Since we could not discount glaucoma, we recommended a full glaucoma work-up to include disc OCT, gonioscopy and pachymetry. To confirm the diagnosis of RP, we ordered a full-field electroretinogram (ERG) and genetic testing. We used Invitae’s (Spark Therapeutics) inherited retinal disease genetic testing program that tests approximately 250 genes for variants known to cause inherited retinal disease.26
The ERG revealed diminished a- and b-waves in the both eyes. Genetic testing confirmed phosphodiesterase 6A (PDE6A) gene mutation. PDE6A is a critical component of the visual cycle involved in regulating levels of intracellular cyclic guanosine monophosphate (cGMP) during phototransduction; in RP, low levels of this gene are implicated as the cause of rod apoptosis.3
Results of the ERG and genetic testing established the diagnosis of autosomal recessive RP. We began treatment with oral acetazolamide 250mg BID to treat the CME, as well as brimonidine 0.2% BID OU to lower IOP.
Genetic Testing
Once a tentative diagnosis is established, genetic testing can help confirm the diagnosis, determine the inheritance pattern, and identify at-risk family members. Multi-gene testing is preferred, as it covers a wide-range of gene mutations.7 Several commercial laboratories offer comprehensive retinal dystrophy panels including: Spark Therapeutics, Blueprint Genetics, Molecular Vision Laboratory and Prevention Genetics. Using Spark Therapeutics, we identified our patient as having the PDE6a mutation causing AR-RP.
Patients who have undergone genetic testing should then be referred for genetic counseling. In addition, we recommended the My Retina Tracker Registry—a research database of the Foundation Fighting Blindness. The registry provides more than 20 retinal degenerative diseases, including RP.27 The registry is designed to share information of rare retinal diseases in order to identify individuals who might be interested in participating in research studies and clinical trials.
While we have yet to discover a cure for RP, the field of restorative vision therapy is dynamic. Progress is being made in retinal prostheses as well as in optogenetics, stems cells and gene therapy. We look forward to advances in technology and offering our patients new therapeutic options in the future.
Dr. DelGiodice practices at Associated Eye Physicians in Clifton, NJ.
Ms. Viray is a fourth-year student at Salus University.
1. National Organization for Rare Disorders. Rare disease database: retinitis pigmentosa. www.rarediseases.org/rare-diseases/retinitis-pigmentosa. Accessed May 5, 2020. 2. Fishman GA. Retinitis pigmentosa. Visual loss. Arch Ophthalmol. 1978;96:1185-8. 3. Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238-249. 4. Chang S, Vaccarella L, Olatunji S, Cebulla C, Christoforidis J. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genomics. 2011;12(4):26-75. 5. Fahim AT, Daiger SP, Weleber RG. Nonsyndromic retinitis pigmentosa overview. GeneReviews. www.ncbi.nlm.nih.gov/books/NBK1417. January 19, 2017. Accessed May 5, 2020. 6. Gabai A, Veritti D, Lanzetta P. Fundus autofluorescence applications in retinal imaging. Indian J Ophthalmol. 2015;63(5):406-15. 7. Duncan JL, Bernstein PS, Birch DG, et al. Recommendations on clinical assessment of patients with inherited retinal degenerations—2016. American Academy of Ophthalmology. www.aao.org/clinical-statement/recommendations-on-clinical-assessment-of-patients. June 2016. Accessed May 5, 2020. 8. Robson AG, Tufail A, Fitzke F, et al. Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina. 2011;31:1670-9. 9. Ogura S, Yasukawa T, Kato A, et al. Wide-field fundus autofluorescence imaging to evaluate retinal function in patients with retinitis pigmentosa. Am J Ophthalmol. 2014;158:1093-8. 10. Liu G, Liu X, Li H, Du Q, Wang F. Optical coherence tomographic analysis of retina in retinitis pigmentosa patients. Ophthalmic Res. 2016;56:111-22. 11. Aizawa S, Mitamura Y, Baba T, et al. Correlation between visual function and photoreceptor inner/outer segment junction in patients with retinitis pigmentosa. Eye (Lond) 2009;23:304-8. 12. Maiti A, Uparkar M, Natarajan S, Borse N, Walinjkar J. Principal components’ analysis of multifocal electroretinogram in retinitis pigmentosa. Indian J Ophthalmol. 2011;59(5):353‐7. 13. Iftikahr M, et al. Progression of Retinitis Pigmentosa on multimodal imaging: The PREP-1 Study. Clin Exp Ophthalmol. 2019;47(5):605-13. 14. Massof RW, Finkelstein D. Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Arch Ophthalmol. 1993;111:751-4. 15. Aleman TS, Duncan JL, Bieber ML, et al. Macular pigment and lutein supplementation in retinitis pigmentosa and Usher syndrome. Invest Ophthalmol Vis Sci. 2001;42(8):1873-81. 16. Bazan NG. Decreased content of docosahexaenoate and arachidonate in plasma phospholipids in Usher’s syndrome. Biochem Biophys Res Commun. 1986;141(2):600-4. 17. Gong J, Rosner B, Rees DG, et al. Plasma docosahexaenoate levels in various genetic forms of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1992;33(9):2596-602. 18. Berson E. Further evaluation of docosahexaenoate in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup anaysis. Arch Ophthalmol. 2004;122(9):1306-14. 19. Berson EL. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128(4):403-11. 20. Marmor MF, Norton EW, Clowes, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(11):1460-1; author reply 1463-5. 21. Jayakody SA, Gonzalez-Cordero A, Ali RR, Pearson RA. Cellular strategies for retinal repair by photoreceptor replacement. Prog Retin Eye Res. 2015;46:31-66. 22. Vingolo EM, Rocco M, Grenga P, et al. Slowing the degenerative process, long lasting effect of hyperbaric oxygen therapy in retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2008;246:93-8. 23. Birch DG, Weleber RG, Duncan JL, et al. Randomized trial of ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for retinitis pigmentosa. Am J Ophthalmol. 2013;156:283-92. 24. Berkrot, B. Spark’s price for Luxturna blindness gene therapy too high: ICER. Reuters. www.reuters.com/article/us-spark-icer/sparks-price-forluxturna-blindness-gene-therapy-too-high-icer-idUSKBN1F1298. January 12, 2018. Accessed May 5, 2020. 25. da Cruz L, Dorn JD, Humayan MS, et al. Five-year safety and performance results from the Argus II retinal prosthesis system clinical trial. Ophthalmology. 2016;123:2248-54. 26. Invitae. ID your IRD. www.invitae.com/en/idyourird. Accessed May 5, 2020. 27. Foundation Fighting Blindness. My retina tracker registry. www.fightingblindness.org/my-retina-tracker-registry. Accessed May 5, 2020. |
