Miller Fisher syndrome (MFS), a rare, self-limiting variant of Guillain-Barré syndrome (GBS), is an anti-GQ1bIgG antibody syndrome that affects the peripheral and central nervous systems.1 An acute neuromuscular polyneuropathic condition, MFS causes an ascending paralysis with a classic presentation triad of ophthalmoplegia, ataxia and areflexia. Researchers first described the clinical triad in 1932.1 Two decades later, Canadian stroke specialist Charles Miller Fisher was the first to publish a report in 1956 that described the clinical findings and defined the condition as a limited form of GBS.2
Symptoms occur over several days, often in winter and spring, typically following a viral infection, and patients often experience diplopia as one of the first symptoms. When a patient presents with symptoms such as intermittent esotropia, impaired horizontal pursuits and fixed mydriatic pupils, and your neurological exam is abnormal, it’s time to think of GBS, specifically MFS. The following case illustrates the relevant signs and symptoms and reflects the typical outcome.
History
A 27-year-old Caucasian male presented to the VA Eye Clinic during the spring months reporting acute onset binocular diplopia that started one day prior. The patient stated it was constant but only occurred at distance viewing with objects displaced horizontally. There was no eye pain or blurred vision associated with the diplopia. He also complained of recent dizziness and balance difficulties. He reported a sinus infection with significant paranasal congestion that started one week prior to the double vision. Previous ocular and medical histories were unremarkable; the patient was not currently taking any medications and did not have any medication allergies.
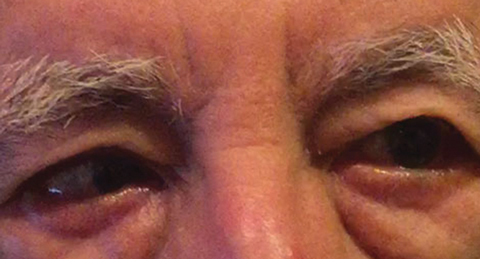 |
| Fig. 1. The differential diagnosis following baseline examination included a partial right CN VI palsy, which could indicate a vascular condition, as seen here in another patient. Click image to enlarge. Photo: Michael DelGiodice, OD |
Diagnostic Data
On baseline examination, uncorrected visual acuities were 20/20-2 OD and 20/25-2 OS. Pupils were 5mm equally round and reactive to light with no afferent pupillary defect. Extraocular motilities were full, smooth and accurate in both eyes with no restrictions. Cover test at distance revealed an intermittent four to six prism diopter right esotropia in primary gaze occurring approximately 50% of the time. Performing cover test at near revealed a four prism diopter esophoria in primary gaze with no tropia.
While not undertaken in this case, clinicians should cover test in primary, right and left gaze. In patients with diplopia, cranial nerve six (CN VI) palsy will manifest as non- comitant deviation, worse in right or left gaze, depending on what side the palsy is on. A decompensating phoria will, on the other hand, show comitant deviation.
Refractive error was -0.50D sphere OD and -0.75D sphere OS and vision was correctable to 20/20 in each eye. Intraocular pressures were within normal limits in both eyes. Anterior and posterior segment findings were unremarkable in the right and left eyes. The optic nerves appeared healthy with a cup-to-disc ratio of 0.15 in both eyes.
Differential diagnoses included a partial right CN VI palsy (Figure 1), myasthenia gravis, a compressive process and a decompensating distance esophoria causing an intermittent right esotropia. The patient denied any previous history of binocular diplopia.
Imaging of the brain and orbits was ordered by the eye clinic to rule out brain involvement, including a compressive lesion. Computed tomography (CT) scans of the brain and orbits were performed and interpreted by the staff neuroradiologist. CT was chosen instead of magnetic resonance imaging (MRI) due to timing and availability; testing and results could be obtained the same day with CT in the clinic after hours. The results were unremarkable with no signs of intracranial hemorrhage, compressive lesion or acute infarction.
CT of the paranasal sinuses revealed moderate to severe chronic paranasal sinus disease and complete opacification of the right maxillary sinus. There was no extension into the orbit. The patient’s family physician was informed of the patient’s symptoms, eye exam findings and CT imaging results and started treatment for maxillary sinusitis with an oral azithromycin five-day dose pack and instructed the patient to follow up with the eye clinic.
The patient returned five days later after completing his oral azithromycin course and stated he experienced no improvement in the distance diplopia; in addition, he experienced the onset of significant photophobia and periocular pain around both eyes. The patient stated that his gait imbalance and walking difficulties were also worsening. On examination, best-corrected visual acuities remained 20/20 in the right and left eyes. Pupils were dilated at 8.5mm with no reactivity to light in either eye (Figure 2). Extraocular motilities revealed lateral restriction with pain upon movement in the right eye and medial restriction with pain upon movement in the left eye consistent with a right gaze palsy (Figure 3). Horizontal and vertical pursuits were saccadic with inaccurate and jerky movements. Anterior and posterior segment findings remained unremarkable OU.
Differential diagnoses after ophthalmologic examination included left internuclear ophthalmoplegia, right gaze palsy and right gaze palsy with left internuclear ophthalmoplegia, which could account for extraocular motility findings. Other differentials include a midbrain lesion or myasthenia gravis. Differential diagnoses for pupil dilation include third nerve (CN III) palsy, trauma and pharmacologic dilation. The patient did not have any other findings consistent with a third nerve palsy and denied trauma or exposure to pharmacological agents.
The patient was referred to neurology for further assessment. Neurological examination reported the patient was fully alert and oriented with no loss of facial sensation. The patient exhibited mild ataxia with difficulty walking in tandem and standing from a squatting position. Proximal weakness in both the upper and lower extremities and abnormal reflexes were present. All sensory modalities were intact. MRI and magnetic resonance angiography (MRA) of the brain and orbits were unremarkable. Cerebrospinal fluid protein was elevated at 75mg/dL (reference range: 15mg/dL to 45mg/dL) with no other abnormalities. Electromyogram testing was unremarkable and showed no abnormalities in nerve conduction from the spine to the feet and hands.
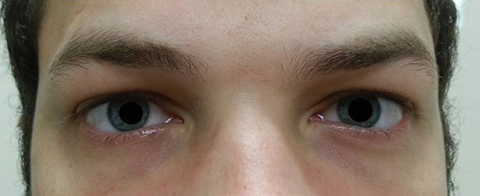 |
| Fig. 2. The patient’s pupils were dilated 8.5mm with no reactivity in the right or left eyes. Click image to enlarge. |
Diagnosis
The patient presented with acute symptoms of diplopia, ataxia and areflexia. Given the intermittent right esotropia, impaired horizontal pursuits, fixed mydriatic pupil, mild ataxia and abnormal reflexes on neurological exam and elevated cerebrospinal fluid protein, the diagnosis MFS was made. The patient was admitted to the hospital to monitor for any further complications. An eye patch was provided to the patient to cover the right eye as needed for diplopia.
No published diagnostic criteria for Miller Fisher syndrome currently exists. The diagnosis is usually made through the presentation of the clinical triad along with imaging and cerebrospinal fluid studies. MRI is typically unremarkable in the condition, although some studies have shown central lesions in the midbrain, pons and lower medulla. Electrophysiological studies show reduced or abnormal peripheral sensory conduction.17
Cerebrospinal fluid protein is often elevated with no other abnormal findings. In a landmark study, 64.4% of MFS patients showed elevated spinal fluid protein.8 A positive anti-GQ1b IgG antibody test allows for a definitive diagnosis. In one study, anti-GQ1b IgG antibody was present in up to 95% of patients with the condition and absent in controls.3
The neurologist diagnosed the patient with MFS based on the clinical triad of ophthalmoplegia, ataxia and areflexia, an unremarkable MRI and MRA of the brain and elevated cerebrospinal fluid protein. Therefore, anti-GQ1b IgG antibody testing was not performed in this case.
Several clinical features of MFS are also seen in Guillain-Barré syndrome and Bickerstaff’s brainstem encephalitis, making the diagnosis challenging. Patients with only Guillain-Barré present with limb weakness, sensory loss, cranial neuropathy and areflexia with no ophthalmological manifestations. Patients with Bickerstaff’s brainstem encephalitis typically present with ophthalmoplegia, ataxia, hyper-reflexia and a disturbed consciousness. Since several symptoms and signs are present in all three conditions, anti-GQ1b IgG antibody titer testing is helpful for a more definitive diagnosis. Anti-GQ1b IgG antibody is positive in a large majority of Miller Fisher syndrome patients compared with 66% in Bickerstaff’s brainstem encephalitis and 26% of Guillain-Barré syndrome patients.16
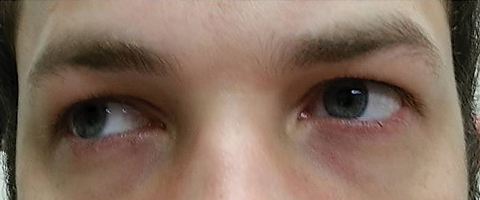 |
| Fig. 3. Extraocular motilities revealed restriction in dextroversion. Click image to enlarge. |
Treatment and Follow-up
The patient was evaluated the day after admission, when he stated he experienced no visual changes or improvement in balance. Binocular diplopia was still constant; therefore, the right eye remained patched. Eye pain with far gazes and fast eye movements were still present. On limited bedside examination, best-corrected visual acuities remained 20/20 in the right and left eyes. Pupils were dilated at 8.0mm in both eyes with minimal reactivity, which was slightly improved from the day prior. Color vision was normal in the right and left eyes. Extraocular motilities were full in all quadrants with pain still present on far horizontal gazes. Horizontal pursuits remained saccadic with jerky movements and endpoint nystagmus. The optic nerves appeared healthy with distinct margins and a cup to disc ratio of 0.15 in both eyes.
Two days after, the patient stated an improvement in all symptoms and that his eyes “feel better today than they have been in a while.” Eye pain and binocular diplopia were both resolved. Visual acuities remained 20/20 in the right and left eyes. Pupil sizes were 7.5mm in dark and 7.0mm in light with improved direct and consensual responses in both eyes. Extraocular motilities were full in all quadrants with no eye pain. Horizontal pursuits remained saccadic with jerky movements and endpoint nystagmus. Neurological exam revealed abnormal reflexes, which remained stable. The treatment options for MFS include plasmapheresis and intravenous (IV) immunoglobulin IgG, but these were withheld given the improvement in ophthalmoplegia and ataxia.
Four days later, the patient reported all symptoms were still improving, with some difficulty focusing and tracking while using the computer. Balance was 80% resolved and there were few occurrences of distance diplopia. Acuities remained 20/20 OU. Pupils were 5.5mm in dark and 4.0mm in light with 3+ direct and consensual responses OU. Extraocular motilities were full in all quadrants with no eye pain. Horizontal pursuits were improved, with no saccadic movements. Monocular accommodation was normal, which was measured 8.5 and 9 prism diopters OD and OS, respectively.
One week later, the patient reported his vision returned to near normal with no diplopia and mild difficulties focusing and tracking. Balance was 100% resolved. Visual acuities remained 20/20 in the right and left eyes. Pupil sizes were 5.5mm in dark, 4.0mm in light with 4+ direct and consensual responses OU. Extraocular motilities were full in all quadrants with no eye pain. Horizontal pursuits improved with no observed saccadic movements. Ocular health was unremarkable OU.
Treatment was withheld given the resolution of ophthalmoplegia and ataxia with stable areflexia. Full recovery of opthalmoplegia and ataxia took two weeks. Management included close monitoring and follow up in four weeks; the patient was advised to return sooner if any of his symptoms returned.
Clinical Features
In well-described studies of MFS, all patients presented with the clinical triad of ophthalmoplegia, ataxia and areflexia.4,5 Onset of symptoms typically occur over several days, and patients suffer a viral infection one to four weeks prior to the onset of clinical symptoms.4,5 According to one study of 50 patients with the condition, the mean interval between infection onset and development of neurological symptoms is eight days.4 Other signs of MFS include slurred speech, difficulty swallowing and an abnormal facial expression with an inability to smile or whistle.
Ophthalmological features. Practitioners should be on the lookout for mydriasis, lid retraction, acute angle closure and diplopia secondary to nerve palsies affecting CN III, IV and VI.3,8,10,22,24 Diplopia is the most common ophthalmological symptom reported in many MFS studies.8,16,25,26,27 Diplopia is also the initial symptom reported in 38.6% of 223 patients in one landmark study and 65% of 466 patients in a second study.8,27 In the second study, 100% of patients exhibited external ophthalmoplegia, while internal ophthalmoplegia was present in 35% of patients.27 External ophthalmoplegia refers to impairment of external extraocular muscles. Internal ophthalmoplegia refers to impairment of the pupillary sphincter and ciliary muscle. Complete ophthalmoplegia affects both external and internal muscles.
In a retrospective study of 19 patients with MFS, all presented with one or more nerve palsies affecting the extraocular muscles.25 Other studies show MFS patients may experience multiple cranial nerve palsies, which can manifest bilaterally.22,24,25 One common finding found among anti-GQ1b IgG positive disorders is abduction deficit consistent with a unilateral or bilateral CN VI palsy.26,28 Internal ophthalmoplegia causes pupil mydriasis, where pupil constriction to light and/or near stimulation can range from minimal to absent.8,26 In one report, mydriasis was present in 42% of patients. Involvement of CN VII occurred in approximately 45.7% of MFS patients causing facial weakness, inability to smile or whistle.8
Ataxia. The cause of this clinical feature in MFS is not fully understood. Debate exists on whether ataxia is caused by dysfunction centrally in the cerebellum or peripherally. The first hypothesis, proposed by Dr. Fisher, postulated the involvement of Ia-afferent neurons.2 This was supported by a second study decades later, which showed abnormalities of Ia-afferent fibers in MFS.11 Abnormal peripheral nerve involvement has also been proposed with a mismatch between proprioceptive input from muscle spindles and kinesthetic information from joint receptors.12
There has also been supporting evidence that ataxia originates in the cerebellum. A predominance of anti-GQ1b IgG antibody in the cerebellum was found using immunocytochemical staining of the human cerebellum and further confirmed with western blot studies, which showed increased anti-GQ1b IgG antibody in MFS patients compared with control, although the mechanism for cerebellar involvement has not been studied.13,14 In 2015, researchers published a case report of a MFS patient who underwent MRI. There was a reduced N-acetylaspartate/creatine ratio, suggesting cerebellar dysfunction, which returned to normal 2.5 months following recovery.23
Areflexia. This clinical feature, along with depression of deep tendon reflexes, is a sign of peripheral nervous system involvement in MFS. Electrophysiological studies have validated abnormal peripheral nerve conduction in GBS and MFS.3 In one study, 81.6% of MFS cases presented with arefexia.8 Another landmark study showed all patients experienced areflexia, which was still present six months after onset.4
Just How Common is MFS?Epidemiological data on Miller Fisher syndrome is limited. The incidence of MFS is quite rare, occurring in 0.09 per 100,000 people annually.3 MFS occurs more commonly in patients with Guillain-Barré syndrome, from 3% to 25%.4,6,7 This association occurs more in patients of Far East descent, suggesting a possible genetic component to MFS. The mean age of onset is 43, with a range of 13 to 78.5 MFS affects males twice as much as females with a ratio of two to 1.03.5 MFS occurs more in the winter and spring seasons.5,6,8 The exact cause of the seasonal predilection has not been determined, but is likely associated with the post-infectious nature of the condition as bacterial and viral infections have been found to trigger an autoimmune response causing production of the anti-GQ1b IgG antibody in MFS. |
Pathophysiology
The pathophysiology of Miller Fisher syndrome is not well understood; however, several hypotheses exist from immunological and histological studies. It is well known that the condition lies within the spectrum of anti-GQ1b IgG antibody syndromes along with Guillain-Barré syndrome and Bickerstaff’s brainstem encephalitis.
The ganglioside GQ1b is a group of complex lipids involved in the central and peripheral nervous systems. GQ1b is a component of the plasma membrane structure in cranial nerves that supply the extraocular muscles and is involved in cell function at the presynaptic neuromuscular junction.
An autoimmune mechanism from a preceding triggering infection will produce the anti-GQ1b IgG antibody, which damages ganglioside GQ1b function, causing demyelination. Histological studies have shown demyelination and axonal swelling in peripheral and oculomotor cranial nerves.15 The anti-GQ1bIgG antibody is absent in healthy patients, but positive in over 90% of patients with MFS.16,25
Bacterial and viral infections have been found to trigger an autoimmune response, causing production of the anti-GQ 1b IgG antibody. The following infective agents that have been associated with MFS include mycoplasma pneumonia, HIV, Campylobacter jejuni, Hemophilus influenza, Helicobacter pylori and Epstein-Barr virus.4,16 In one report that looked at 466 MFS patients, 90% had an antecedent illness, including upper respiratory infection, diarrhea or both.27
Prognosis and Treatment
Miller Fisher syndrome is a self-limiting disease and has an overall positive prognosis. Symptoms generally improve after a few weeks with full recovery typically occurring in two to three months. In one study, recovery started at a median of 13 days from symptom onset, and complete resolution of ophthalmoplegia and ataxia occurred in six months.4,5 Relapses have been found to occur in less than 3% of cases.4,5
Although MFS is typically self-limiting, systemic complications have been associated with the condition. Upper respiratory infections have been found in 56% to 76% of MFS patients, which can progress to respiratory failure requiring mechanical ventilation.4,18,19 Other rare serious complications include cardiomyopathy, lactic acidosis and coma.3,20
The treatment options for MFS include plasmapheresis, IV immunoglobulin IgG, and monitoring without treatment. Researchers postulate that plasmapheresis and IV IgG may be effective to speed resolution time since antibody Gq1b is IgG in class and the half-life of IgG is approximately 21 days, which is longer than the five to six day half-lives of IgM and IgA.19 However, no randomized double-blinded placebo-controlled studies investigating these treatments have been conducted.
One retrospective study failed to show plasmapheresis changed the chance of full recovery and the time to resolve ataxia and ophthalmoplegia; further, the interval from onset to start of plasmapheresis treatment did not affect time to resolution or the severity of symptoms at onset. However, one case report indicates that plasmapheresis is indicated when profound ataxia, motor and respiratory impairment is present.21
Patients should be monitored closely for risk of developing serious systemic complications such as upper respiratory failure. Studies show no significant improvement in recovery time with IV IgG or plasmapheresis treatment in MFS, so monitoring may be a practitioner’s best option.
While rare, the condition need not go undiagnosed. By recognizing the key clinical features, practitioners can reveal the cause of their patient’s symptoms and gain confidence in a full recovery.
Drs. Wang and Cantrell are staff optometrists at the Orlando VA Medical Center. Dr. Cali is a staff optometrist at the Lee County VA Clinic. The authors wish to thank Joseph Miller, OD, Paul Gruosso, OD, and Vanessa Santos, OD, for assistance with this manuscript.
1. Collier J. Peripheral neuritis: The Morisa lectures. Edinburgh Med J. 1932:39:601-18. |
