 |
An eight-year-old Asian male was referred to our clinic for unexplained vision loss in both eyes. Starting about five months earlier, his parents stated, he began sitting closer to the board at school and pulling books in closer to read. They noted he was also having some trouble differentiating colors.
His overall health was unremarkable. He was not taking any medications, nor did he have any medication allergies. His family medical and ocular history was non-contributory.
Upon examination, his visual acuity measured 20/100 OU with no improvement after mild hyperopic refractive error was trialed. Extraocular motilities showed full range of motion. His confrontation fields were full to careful finger counting in both eyes. Pupils were round and reactive to light with no afferent defect. He was able to see the test plate on color vision, but scored 0/10 in each eye.
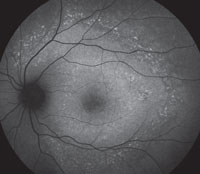
Slit-lamp examination of the anterior segment yielded nothing remarkable. Posterior segment examination revealed optic nerves with moderate, physiologic cupping and mild temporal pallor, in his right eye more than in his left. The fundus photos depict what was observed clinically (Figures 1a, 1b and 1c). Fundus autofluorescence (FAF) (Figures 2a and 2b) and intravenous fluorescein angiography (IVFA) (Figure 3) were performed and is available for review.
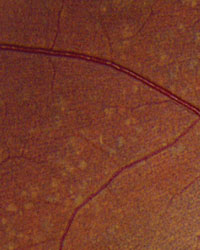 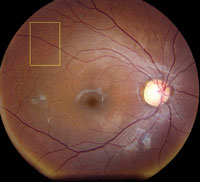 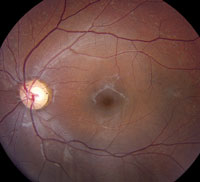 | |
| Fig. 1a-c. What do the fundus images tell you about our eight-year-old Asian male patient? Take special note of the magnified area at the top. |
Take the Quiz
1. What do the discolored lesions in both eyes’ fundus photos represent, and where are they located?
a. Lack of melanin granules; within the RPE.
b. Drusen; under Bruch’s membrane.
c. Lipofuscin; between RPE and photoreceptors.
d. Lipofuscin; within the RPE.
2. On FAF, how would one describe the pattern seen?
a. Foveal hypoautofluorescence with punctate hyperautofluorescent lesions.
b. Scattered window defects with normal autofluorescence.
c. Unremarkable.
d. Normal macular autofluorescence with peripheral lesions.
3. What is the likely diagnosis?
a. Progressive cone dystrophy.
b. Stargardt’s macular dystrophy.
c. Bietti’s crystalline dystrophy.
d. North Carolina macular dystrophy.
4. Which two features does the IVFA for our patient’s condition typically manifest?
a. Late leakage near fovea from leaky vessels.
b. Silent choroid from RPE blockage defect.
c. Early punctate hypofluorescence surrounding arcades
d. Late staining of retinal flecks.
Discussion
Our patient had reduced central vision, a silent choroid on flurorescein angiography, and an early bulls-eye pattern on fundus autofluorescence. Though he lacked the stereotypical “pisciform” or “tri-radiate” retinal flecks, his diagnosis was most consistent with autosomal recessive Stargardt’s macular dystrophy (or Stargardt’s disease).
Stargardt’s disease is the most commonly inherited childhood autosomal recessive retinal dystrophy. Less commonly, some Stargardt’s disease mutations have an autosomal dominant transmission.1 The genes typically affected are ABCA4, ELOVL4 or PROM1. This condition affects the retinal pigment epithelium (RPE) and photoreceptor layer and typically has an onset in childhood or young adulthood. Some patients can develop symptoms of visual acuity loss as late as the fourth or fifth decade of life.
Diagnosis of Stargardt’s disease can be straightforward in the event that the patient presents in such a fashion. However, there is variability in the shape/formation or even presence of the retinal flecks, the age of onset, the decline in visual acuity, and the OCT findings—all of which can complicate the diagnosis.
Clinical testing for this condition can be extremely helpful, especially because vision can be reduced before any retinal signs are present. Recent research suggests one of the earliest signs is an abnormally thickened external limiting membrane (ELM) on OCT.2 The ELM is located between the photoreceptor cell bodies (outer nuclear layer) and the photoreceptor inner segments, and its thickening indicates early photoreceptor damage. As the disease progresses, OCT reveals a clear dropout of the outer retinal layers, as seen in our patient.
The FAF lights up in the areas where there is loss of RPE and overlying photoreceptors on OCT. The areas of hyperautofluorescence relate to areas where there is still functioning, but defective, RPE. The overall area of abnormal FAF has been found most specifically to relate to IS/OS junction loss that may extends beyond the damaged retina visible on direct observation.3
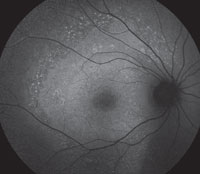  | |
| Figs. 2a and 2b. Can you use these FAF images to explain the patient’s symptoms? |
IVFA may not be necessary for diagnosis, but if performed, shows a key feature: the “silent choroid.” The absence of choroidal flush is not actually a problem with the choroid. The hypofluorescence seen is due to signal blocking from high-grade lipofuscin accumulation within the RPE lying above. Foveal patches of hyperfluorescence are window defects with increased signal transmission through defective RPE. The flecks themselves present as early blockage and late, non-progressive staining.4 Frequently, a bulls-eye pattern will be present as well.
Unfortunately, there is no current treatment for Stargardt’s disease. There are, however, ongoing studies in early phases for deuterium enrichment of vitamin A to slow lipofuscin formation and storage, subretinal transplantation of stem cell-derived RPE cells and gene therapy.
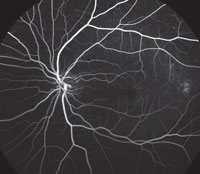 | |
| Fig. 3. This is the venous phase of the fluorescein angiogram. What does it reveal about our patient’s condition? |
Handling progressive vision loss in a child is a difficult topic to discuss, so both our patient and his parents were carefully counseled about his condition. We discussed future prognosis and assistance programs for school. He is not currently enrolled in any studies, but may be a candidate in the future at our facility.
This case was written by Alison Bozung, OD, optometric resident at Bascom Palmer Eye Institute.
1. Agarwal A. Gass’ Atlas of Macular Disease. 5th ed. Waltham, MA: Elsevier Saunders; 2012. 278-85.2. Burke T, Yzer S, Zernant J, et al. Abnormality in the external limiting membrane in early Stargardt disease. Invest Ophthalmol Vis Sci. 2014 Oct;55(10):6139-49.
3. Gomes N, Greenstein V, Carlson J, et al. Comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009 August;50(8):3953–9.
4. Agbaga M, Tam B, Wong J, et al. Mutant ELOVL4 that causes autosomal dominant stargardt-3 macular dystrophy is misrouted to rod outer segment disks. Invest Ophthalmol Vis Sci. 2014 Jun; 55(6): 3669–80.
Retina Quiz Answers:
1) d; 2) a; 3) b; 4) b & d.
