 |
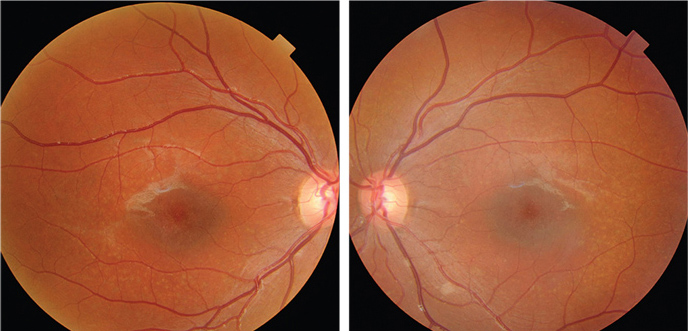
| Misdiagnosis of Stargardt’s disease as AMD should be avoided to prevent futile invasive treatments with potential complications. Photo: Jerome Sherman, OD. Click image to enlarge. |
As it’s among the more rare retinal conditions, Stargardt’s disease patients are often misdiagnosed. More knowledge of its late-onset subtype is necessary to avoid misdiagnoses with diseases with similar presentation such as dry AMD with geographic atrophy. Researchers based in the Netherlands have described the phenotypic appearance, the disease progression and the genetic background of a uniquely large cohort of late-onset Stargardt’s type 1 patients. They found that those with this specific subtype most commonly harbor one severe and one mild ABCA4 variant. The general patient also presents with typical fundus flecks and develops retinal atrophy in a foveal sparing pattern, resulting in a preserved central vision.
This study, published in Ophthalmology, reviewed clinical data including age of onset, initial symptoms and best-corrected visual acuity of 71 patients with late-onset Stargardt’s. The mean age was 55.0 years. The team performed quantitative and qualitative assessment of retinal pigment epithelium atrophy (RPE) on fundus autofluorescence (FAF) images and optical-coherence tomography (OCT) scans.
A combination of a mild and severe variant in ABCA4 was the most found genotype (n = 49; 69.0%). The most frequent allele c.5603A>T (p.Asn1868Ile) was present in 43 of 71 patients (60.6%). No combination of two severe variants was found. All patients presented with flecks on FAF imaging at first presentation. Foveal sparing atrophy was present in 33.3% of the eyes, while atrophy with foveal involvement was found in 21.1%. Extrafoveal atrophy was present in 38.9% of the eyes, and no atrophy in 6.7%.
Time-to-event curves showed a median duration of 15.4 years from onset to foveal involvement. The median visual acuity decline was -0.03 Snellen decimals per year. Median atrophy growth was 0.590mm2/year for definitely decreased autofluorescence and 0.650mm2/year for total decreased autofluorescence.
“This suggests that atrophic lesions in the late-onset disease subtype grow more outwards than centrally, leaving the fovea intact longer,” the researchers wrote in their paper. “While the majority of the late-onset patients have well-demarcated RPE atrophy, a third present with mottled atrophy, i.e., poorly demarcated questionably decreased autofluorescent atrophy.”
“Retinal changes in late-onset Stargardt disease typically start parafoveal and may not cause noticeable symptoms initially,” the team noted. “Patients only seek medical attention after they start experiencing complaints. This makes it challenging to determine the exact age of disease onset.”
“The use of multimodal imaging, consisting of color fundus photography, FAF and OCT, is sufficient in distinguishing between these two diseases, rendering the need for more invasive angiography techniques unnecessary,” the study concluded. “To the trained eye, unique features can be recognized but differentiation solely based on clinical phenotype can remain difficult and genetic analysis of the ABCA4 gene will ultimately tell the difference.”
Li CHZ, Pas JAAH, Corradi Z, et al. Study of late-onset Stargardt type 1 disease: characteristics, genetics and progression. Ophthalmology. August 18, 2023. [Epub ahead of print]. |
