 | |
|
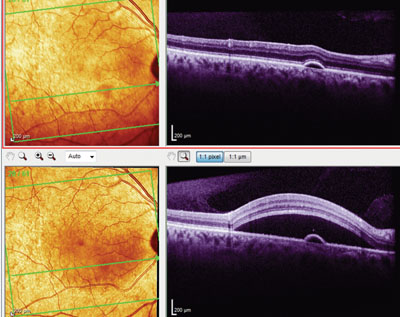
OCT image of the right eye at initial presentation (top) and first follow-up visit (bottom) shows a pigment epithelial detachment. Note the movement of the focal neurosensory detachment from a superonasal macular location (top) to a more central macular location (bottom).
|
When I first saw her, she described a cloud-like fog in the vision of her right eye for the past two weeks. She also reported a ‘spot’ in her vision in the right eye, which had gotten bigger during the two weeks. She also complained of increasing photophobia (OS>OD), which began about the same time.
Diagnostic Data
At this initial visit, her visual acuity was 20/50 OD and 20/30 OS, with pinhole acuity of 20/40- OD and 20/20- OS. Pupils were normal with no afferent defect. Confrontation visual fields were remarkable for a central depression (OD>OS).
Slit-lamp exam showed grade 3+ cells and 2+ flare in the anterior segments OU. No keratic precipitates, iris nodules or synechiae were noted in either eye. The limbal episclera was moderately injected OU. Intraocular pressure measured 20mm Hg OU at 3:45 p.m.
Upon dilation, her crystalline lenses were clear OU. The anterior and posterior vitreous evaluations OU showed no evidence of cells. Stereoscopic disc evaluation demonstrated healthy neuroretinal rims with normal cupping in both eyes.
Macular evaluation of the right eye revealed a large area (approximately four disc diameters) of retinal thickening located adjacent to the optic nerve, just inferior to the superior temporal retinal arcades. The inferior aspect of this retinal thickening extended to the superior aspect of the foveal avascular zone. The left macula was unremarkable, as were the peripheral retinas in both eyes.
Optical coherence tomography evaluation of the right macula revealed a large neurosensory retinal separation in the area of the retinal thickening, as well as a small pigment epithelial detachment (PED) in the inferior macula. The focal neurosensory retinal detachment was not centered in the macula, but was structurally characteristic of an atypical case of central serous retinopathy. Interestingly, the macular OCT of the left eye also demonstrated a small PED inferonasal to the foveal avascular zone.
Diagnosis and Management
Based upon the clinical presentation, I diagnosed atypical central serous retinopathy (CSR) OD, bilateral juxtafoveal PEDs OU and anterior uveitis OU. Given the bilaterality of the anterior uveitis and PEDs, I had to consider the possibility of a systemic etiology. Accordingly, I ordered the following initial labs: CBC with differential, ANA, RA latex, ESR, CRP and ACE levels.
To quiet the anterior chamber reactions, I prescribed atropine 1% BID OU, Pred Forte (prednisolone acetate 1%, Allergan) Q4H OU and Acular LS (ketorolac tromethamine 0.4%, Allergan) QID OU.
The patient returned in four days with slightly worsening vision in the right eye, and stabilization of the photophobia OU. She reported that she was compliant with her drops.
Pinhole acuity was unchanged OU. Intraocular pressure at this visit was 21mm Hg OD and 20mm Hg OS. Her anterior chambers were unchanged, still showing notable cells and flare OU. No synechiae were noted in either eye.
Her retinal evaluations, both at the slit lamp and via OCT imaging, demonstrated a gross inferior billowing shift of the focal neurosensory retinal detachment to a more central macular location, now encompassing the pre-existent PED. The PED in the left eye increased retinal thickness by 20μm. All of her lab findings were normal, except for a slightly elevated CRP level of 6.8mg/L.
Over the next several weeks, the patient remained moderately compliant with her medication schedule, although she did report on two occasions that she accelerated her steroid tapering “as my eyes felt better.” At one of the follow-up visits after she decreased her dosage of steroids, her anterior chambers had re-flared, as had the CSR OD, which previously begun to clear. As a result, I directed her to increase the topical steroids to the initially prescribed level.
Unfortunately, at about this same time, IOP in both eyes began to increase. I prescribed timolol 0.5% QAM OU, and her IOP in both eyes soon dropped from the mid-30s to the upper teens.
By July 2013, her retinas were clear with no evidence of PEDs or CSR. Further labs, including thyroid function studies and chest films, were negative. Histo and Lyme titers were also normal. The anterior chambers were relatively quiet with trace cells and flare. I told her to gradually taper the steroids, with an anticipated cessation in four to six weeks. I kept her on timolol 0.5% QAM OU, but adjusted Acular to QD OU.
Unfortunately, on follow-up in August 2013, she reported increased periocular discomfort and decreased vision. She also said that the timolol was irritating her eyes, so she discontinued that drop OU. At this visit, best-corrected visual acuity was 20/20 OU. Anterior chambers showed 1+ cells and flare, and IOP had risen to 48mm Hg OD and 51mm Hg OS. I prescribed Simbrinza (brinzolamide1%/brimonidine 0.2%, Alcon) BID OU.
At the patient’s most recent visit in early November 2013, her anterior uveitis had cleared (and has remained so for 30 days), and the CSR and PEDs also remained clear OU. IOP was 18mm Hg OD and 21mm Hg OS. The patient is now medicated OU with Acular QD, Pred Forte BID, Simbrinza BID and timolol 0.5% QAM.
Our current plan: a very slow taper of topical steroids to prevent re-flare, while continuing the IOP-lowering medications to control the steroid-induced glaucoma.
Discussion
This case presents a challenge: How can we control the anterior uveitis and retinal conditions while managing the steroid-induced IOP increase?
Keep in mind that when topical steroids are used to treat anterior uveitis, an IOP rise might occur for one of two reasons. First, the IOP rise may be due to the use of the steroid itself, which is believed to increase glycosaminoglycans and reduce outflow in steroid responders. In such cases, the obvious treatment is to lower or eliminate the topical steroids.
On the other hand, an IOP rise may occur due to inflammation and inflammatory debris (originating in the anterior chamber) that clogs the trabecular meshwork. When the inflammation itself is the root of the IOP rise, topical steroids may actually need to be increased to ultimately result in lower IOP (by reducing trabecular inflammation).
Trabeculitis is nearly impossible to visualize gonioscopically. However, inflammatory debris can occasionally be seen on gonioscopy. In this patient’s case, gonioscopy of the anterior chamber showed open angles with no discernable debris. Could the trabeculum be inflamed, causing her elevated IOP? Or could she simply be a steroid responder? In this case, the answer to both questions is “yes.” I concluded that she was a true steroid responder because the anterior uveitis was not murky or granulomatous in appearance, which is more likely to occur with trabecular inflammation.
Managing a steroid responder is rather straightforward. While we should ideally use steroids at a minimum, it’s really the inflammatory condition that dictates the steroid dosage. Consequently, elevated IOP can be reduced by a variety of pressure-lowering agents. The key is to lower IOP and keep it lowered during the duration of the elevated IOP—whether that’s for days or for years.
