 |
History
A 44-year-old Caucasian male reported to the office with a chief complaint of poor night vision. He explained that he’d seen other eye doctors and that they had told him he had some “freckles” in his left eye. His systemic and ocular histories were unremarkable, and he denied allergies of any kind.
Diagnostic Data
His best-corrected entering visual acuities were 20/20 OU at distance and near. His external examination was normal, with evidence of sluggish pupil on the left side. His peripheral confrontation visual field was distorted and constricted in the left eye. The biomicroscopic examination of the anterior segments found normal structures with Goldmann applanation pressures measuring 15mm Hg OU. The pertinent dilated fundus findings are demonstrated in the photograph.
Your Diagnosis
Does the patient’s case require any additional tests, history or information? What steps would you take to manage this patient? Based on the information provided, what diagnosis would you make? What is the patient’s most likely prognosis?
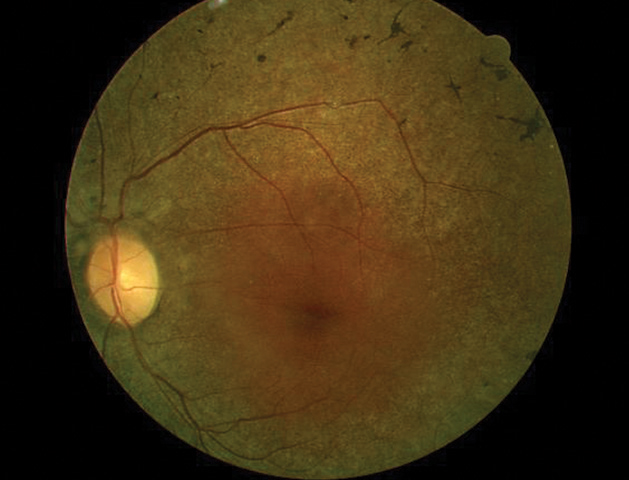 |
| The findings in this fundus photograph portray a patient who presented with self-reported poor night vision. Does the imaging show any signs of disease? |
Discussion
The diagnosis here is retinitis pigmentosa (RP). Additional testing should include central and peripheral threshold perimetry and a referral for electrodiagnostic testing such as electroretinogram (ERG), electroocoulogram (EOG) and dark adaptometry. We ordered cursory bloodwork to rule out systemic sources known to produce pigmentary retinopathy. We also referred the patient to the internist to suggest an electrocardiogram because of documented associations between some pigmentary retinopathies and heart conduction anomalies. Additional history questions we addressed included inquiring if any family member had been diagnosed with a similar problem.
Pathophysiology
RP is a group of inherited disorders affecting 1 in 3,000 to 7,000 people.1,2 It is characterized by abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium (RPE) of the retina, which lead to progressive visual loss.1-14 The predominant symptom is bilateral progressive visual field and acuity loss, often proceeding to blindness.1-13
The pathophysiology of RP is complex.38-46 The common theme of the disease is that it stems from genetic and mitochondrial defects, which produce disturbances in the RPE leading to destruction of the photoreceptors’ outer segment disc membranes.2,8,45 The resultant accumulation of metabolic by-products creates disruption of the normal retinal function, advancing varying combinations of lipofuscin deposition, retinal gliosis, photoreceptor loss, choriocapillaris occlusion, choroidal atrophy and RPE hyperplasia.2,8,44,45 As the RPE alterations progress, the blood-retina barrier becomes eroded, resulting in intraretinal and subretinal leakage.
There are many recognized forms of RP, and while most present with similar findings and outcomes, some presentations are atypical.44 Classification of RP may be made on the basis of inheritance pattern (autosomal dominant, autosomal recessive, X-linked, simplex-no family members, multiplex-multiple genes), age of onset (congenital, childhood onset, juvenile onset, adult onset), predominant photoreceptor involvement (rod-cone, cone-rod), or location of retinal involvement (central, pericentral, sectoral, peripheral).5-13,24,44-47
RP is transmitted through genetic pedigrees echoing all known modes of inheritance.3 To date, 45 causative genes/loci have been identified in non-syndromic RP (for the autosomal dominant, autosomal recessive, X-linked and digenic forms).3 The most common form of RP is a rod-cone dystrophy.3
Syndromic RP is defined as the disease and its variations with associated groupings of signs, symptoms and systemic findings involving one or more organ systems.3,14 The disease process has many variations with the potential for both early or delayed onset.15 Most patients with RP are diagnosed in the second or third decade of life.5-12 Bardet-Biedl syndrome (BBS) and Usher syndrome (US) are the most prevalent syndrome forms involving RP.1,3,7,11,12,16,17 Together they make up almost a quarter of the patients with RP.12 BBS is defined by the association of retinopathy, obesity, hypogonadism, renal dysfunction, postaxial polydactyly and developmental disabilities.1,3,11,12
US is characterized by the combination of congenital or early-onset sensorineural deafness, RP and variable degrees of vestibular dysfunction.11,12,16
Kearns-Sayre syndrome (KS) is a rare disorder consisting of ptosis, limited movement of the eyes and atypical retinal pigmentary changes.7,17
Refsum's syndrome is characterized by defective peroxisomal alpha oxidation of phytanic acid with clinical features that include RP, polyneuropathy, anosmia and hearing loss.12,13,18
Bassen-Kornzweig disease is an autosomal recessive disorder featuring altered lipoprotein metabolism characterized by fat malabsorption, hypocholesterolemia RP, progressive neuropathy and acanthocytosis from early infancy.19,20
Signs and Symptoms
Patients with RP may present with varying symptoms, which are often gradual and insidious with many patients failing to recognize the advancing manifestations until the disease has progressed significantly.1-24 When symptoms are reported, they initially include difficulty with night vision (nyctalopia), vision in bad weather and loss of peripheral vision.1-8,23 Many patients with RP will also experience visually debilitating photopsias as the disorder progresses.21,22 This phenomenon is believed to represent aberrant electrical impulses from the degenerating retina.21 Central visual acuity is generally not affected until very late stages, although variants have been encountered that cause devastating macular compromise early in the disease course (e.g., X-linked recessive RP, RP inversa).8,23,24 Color vision typically remains intact as long as visual acuity is better than 20/40.8,23 Most patients experience their greatest reductions in central vision between the ages of 50 and 80.5-13
The ophthalmoscopic appearance of RP finds attenuation of the retinal arterioles, intraneural retinal pigment (bone spicules) in the midperipheral retina and at perivascular locations, thinning and atrophy of the RPE in the mid and far periphery, preservation of macular integrity (except in RP inversa [macular presentation] and RP sine pigmento), gliotic atrophy of the axons composing the optic nerve (waxy pallor) and choriocapillaris atrophy with increased visibility into the choroid.5-13,24-28 A correlation with acquired optic disc drusen exists in RP.9
In the traditional forms of RP, the appearance and function of the macula and optic nerve remain normal in the early stages of the disease’s development; however, tissue changes in response to the pathology may provoke preretinal gliosis (cellophane maculopathy), which may lead to macular hole, cystoid macular edema and focal RPE defects.29,30 Additional ophthalmologic findings include ectopic lentis, microspherophakia (Weill-Marchesani syndrome), atypical cataract formation, pigment cells in the vitreous, posterior vitreous detachment and associated vitreous hemorrhage.31-34 Most patients with RP are myopic, although high hyperopia has been reported.34-36 A correlation with keratoconus also exists.37
Diagnostics
Electrodiagnostic testing remains the standard for diagnosis.1-3,24,26,38-40 In RP, both the ERG and multifocal electroretinogram (mERG) show significantly diminished red, white, blue and 30hz flicker waves.35,36 The EOG and dark adaptometry remain as staples in diagnosis and monitoring.5-13,39,40 Researchers are also evaluating pupillary light reflex as a new testing modality.41-43 Fundus autofluorescence (FAF) has emerged as a potential tool as well.25,41-43 Genetic testing can determine the risk of expression in offspring and identify specific gene defects in the affected.1-4,9-25
Other clinical entities (syphilis and intestinal polyposis [Gardner’s syndrome]) that cause pigmentary retinopathies must be ruled out with bloodwork.49-52 Serology should be obtained when the diagnosis is unclear or other disorders are suspected.
Serology is used to differentiate syndromic forms of RP. Serum phytanic acid testing is used to rule out Refsum disease, electrocardiogram testing is used to rule classic heart manifestations seen in Kearns-Sayre syndrome and a lipid profile with protein electrophoresis can be used to rule out abetalipoproteinemia.7,12,13,17,18,53
Visual field analysis and electrodiagnostic testing, along with dark adaptometry, should always be obtained to confirm suspected cases.1-3,24,26,36-38 FAF can provide information regarding the integrity of the photoreceptor layer, serving as a secondary instrument for both diagnosis and therapeutic monitoring.41,42
While the suspicion of RP is based upon clinical appearance, many retinopathological conditions mimic its distinctive retinopathy. These may include rubella retinopathy, syphilitic retinopathy, cytomegalovirus retinopathies, toxoplasmosis, cancer-associated retinopathy, pigmented paravenous retinochoroidal atrophy, traumatic retinopathy and retinal drug toxicity secondary to thioridazine, chlorpromazine or chloroquine.48-53
Therapy
There is no known treatment to diminish or reverse the progressive retinal dysfunction in RP.1-53 Management therefore is three-pronged: 1) Prompt diagnosis, 2) Rectify the treatable associated ocular and systemic complications (i.e., refractive error, cataract formation, macular edema, vitreous hemorrhage, hearing loss, dyslipidemia) and 3) Suggest counseling to maintain quality of life.1-53
A pedigree can be done to determine the inheritance pattern and to assess risk to offspring.1-4,14,15,44,46-48 Low-vision services are indicated, as the disorder affects normal visual function.5,36 Field expansion devices, infrared blocking sun lenses and contrast enhancing filters may all be helpful. Visual field analysis and evaluation for cataract development or macular edema should be performed at least biannually.
The artificial silicon retina (ASR) microchip is a new technology designed to be implanted into the subretinal space to treat vision loss.5 The ASR microchip is a 2mm diameter silicon-based device that contains approximately 5,000 microelectrode-tipped microphotodiodes and is powered by incident light.5 Visual function improvements have been documented in patients and included unexpected improvements in retinal areas distant from the implant.5
Animal models have led to the development of therapeutic strategies aimed at identifying and curing specific genetic disorders (gene therapy).1-4,44-47 These include growth factors, calcium blocker applications and vitamin supplements. The use of stem or precursor cells are also being inverstigated.5,6,9,54
The newest treatment options include trophic factor therapy, visual cycle inhibitors and cell transplantation.55 A radically different approach has been given the name neural prosthetics ("artificial vision").55 Rewiring of inner retinal circuits are known to occur naturally in RP, making researchers believe it is possible to create visually useful percepts by stimulating retinal ganglion cells electrically.55 This has lead to the development of techniques to induce photosensitivity in cells that are not normally light sensitive, as well as the development of what is being termed “the bionic retina.”55
Findings in some controlled trials indicate that nutritional interventions, including vitamin A palmitate and omega-3-rich fish, slow disease progression in many patients.56-58 RP Patients placed on vitamin A therapy with docosahexaenoic acid 1,200mg/d demonstrated a slowed disease course over the following two-year period.58 Lutein supplementation of 12mg/d also has shown promise for slowing midperipheral visual field loss in nonsmoking adults with RP taking vitamin A.57
Supplementation therapy is not free of controversy, however. Critics point out there is no universally accepted regimen, and the affects of long-term use remain in question. The literature also suggests that while patients may experience some degree of measurable visual preservation, they do not seem to benefit functionally and must be closely monitored medically while on these preparations.44
|
