 A 42-year-old Hispanic female presented with blurred vision and distortion in both eyes (OS > OD) that persisted for the last 24 hours. She said that her symptoms had started the day before, and were accompanied by a headache. Further, she reported that, “straight lines appeared curved.” The patient said that her headache was better today, but that her vision had not improved.
A 42-year-old Hispanic female presented with blurred vision and distortion in both eyes (OS > OD) that persisted for the last 24 hours. She said that her symptoms had started the day before, and were accompanied by a headache. Further, she reported that, “straight lines appeared curved.” The patient said that her headache was better today, but that her vision had not improved.
Her ocular history was significant for successful soft contact lens wear for the past 24 years. Her medical history was noncontributory. She reported being healthy, and was taking no medications.
On examination, her visual acuity measured 20/25 OD and 20/40 OS. Extraocular motility testing was normal. Confrontation fields were full to careful finger counting OU. Amsler grid testing showed central metamorphopsia involving fixation in both eyes. Her pupils were equally round and reactive, with no evidence of afferent defect. Slit lamp evaluation was significant for trace anterior chamber cells. Her intraocular pressure measured 16mm Hg OU.
On dilated fundus examination, we noted 1+ vitreous cells OU. Optic nerve evaluation showed small cups with good rim coloration and perfusion. We documented significant changes in both maculae and posterior poles (figures 1 and 2).
Additionally, we obtained a fluorescein angiogram (FA) (figures 3 and 4) and an SD-OCT scan (figures 5 and 6).
Take the Retina Quiz
1. What do the changes seen in the maculae and posterior poles represent?
a. Placoid lesions at the level of the retinal pigment epithelium (RPE).
b. Choroidal infarcts.
c. Combined choroidal detachment and retinal detachment.
d. Multiple exudative retinal detachments.
2. What fairly evident clincal findings do the FA and SD-OCT scans reveal?
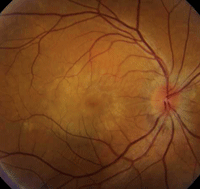
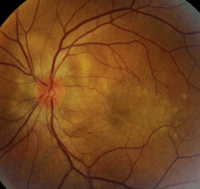
1, 2. Color fundus photographs show obvious retinal changes in the posterior poles and maculae (OD left, OS right).
a. Multiple retinal and RPE detachments.
b. Choroidal detachments.
c. Subretinal mass.
d. Hemorrhagic detachment of the RPE.
3. What is the correct diagnosis?
a. Acute multifocal pigment placoid epitheliopathy.
b. Ocular sarcoidosis.
c. Vogt-Koyanagi-Harada (VKH) disease.
d. Bilateral central serous retinopathy.
4. How should this patient be managed?
a. Observation.
b. Laser photocoagulation.
c. Intravitreal Avastin (bevacizumab, Genentech/Roche) injection.
d. High-dose steroids.
Discussion
It was clear that our patient had serous fluid located in the posterior poles of both eyes. Far less obvious, however, was determining if the fluid represented serous retinal or serous RPE detachments. Both the fluorescein angiogram and the SD-OCT helped us understand the clinical and anatomical changes.
On SD-OCT, we saw that both serous retinal and serous RPE detachments were present in each eye. (The left eye exhibited more fluid and much clearer delineations between the retinal and RPE detachments.) Additionally, the SD-OCT scan revealed localized areas of cystoid macular edema in the right eye.
Although the SD-OCT provided good anatomic perspective, it was the late phases of the FA scan that showed a broader view of our patient’s condition. The delineation of numerous RPE detachments stood out within the mass of diffuse staining throughout the posterior pole and optic nerve.
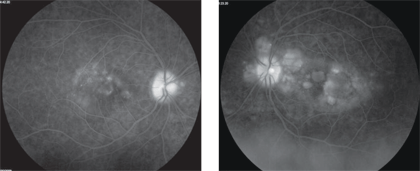
3, 4. Two late-phase FA images show more obvious findings (OD left, OS right).
So, what do these findings represent? Considering her headache the day before, as well as the presence of vitreous cells and numerous exudative retinal detachments in both eyes, it appeared that our patient had VKH disease.
The hallmark finding in patients with VKH disease is bilateral granulomatous uveitis associated with exudative retinal detachments. Several extraocular manifestations also can be present––the most common being cerebrospinal fluid pleocytosis. Other secondary findings include vitiligo, poliosis, alopecia and dysacusis.1 We saw none of those conditions in our patient.
The clinical course of VKH typically follows a pattern that begins with a prodromal stage, in which patients often develop a viral-like illness that lasts three to five days. During this time, patients may have fever, malaise, headaches, dizziness, orbital pain and nausea. Interestingly, with the exception of a headache, our patient did not experience any such symptoms.
Following the prodromal stage, patients move into the uveitic stage. Here, patients typically develop blurred vision (due to exudative retinal detachments), as well as granulomatous inflammation that includes posterior choroid thickening, optic nerve edema, and inflammation of the ciliary body and choroid.
The inflammation also can extend to include anterior chamber cell and flare. We performed an ultrasound on our patient and, indeed, she exhibited posterior choroid thickening.
Next, VKH progresses to the chronic stage, in which patients develop choroid depigmentation. This anatomic change has been referred to as the “setting sun” sign, because the choroid and RPE assume reddish hue that is similar to the color of a beautiful sunset. In this stage, patients also may develop vitiligio and poliosis.
Finally, the chronic recurrent stage of VKH consists of a smoldering panuveitis in which the patient can develop recurrent episodes of granulomatous anterior and posterior uveitis.1
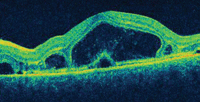
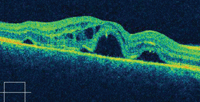
5, 6. The SD-OCT scan shows vertical cuts through both maculae (OD top, OS bottom).
The diagnosis of VKH typically is based on clinical findings. There is some debate on the necessity of ancillary testing, such as lumbar puncture. In cases where the presentation is straightforward, such testing may not be necessary.
However, if the diagnosis is in question, a lumbar puncture may indicate cerebrospinal fluid pleocytosis.
Additionally, human leukocyte antigen (HLA) testing may be useful, depending upon the patient’s ethnicity. For example, VKH is more common in Japanese patients, accounting for 6.8% to 9.2% of all uveitic cases.2 It is also more prevalent in Hispanics, Native Americans and Indians than in whites.
HLA-DR4 testing has been shown to be highly specific in Japanese and Korean patients with VKH. Interestingly, HLA-DR4 and subtype HLA-DR1 were detected in 89% of Mexican mestizo patients living in Southern California who were diagnosed with VKH.1,3 Our patient did not have HLA testing, because the diagnosis was fairly uncomplicated.
The mainstay of treatment is early and aggressive systemic corticosteroid therapy (80mg to 100mg oral prednisone per day), followed by a slow taper over a six-month duration. Alternatively, intravenous methylprednisolone can be administrated for three days, followed by a slow taper of oral prednisone.1
Most VKH patients respond very well to corticosteroid therapy, with rapid symptom improvement that includes complete resolution of the exudative retinal detachments and a restoration of visual function.
We started our patient on 80mg of oral prednisone. Over the course of one month, the fluid resolved and her visual acuity returned to 20/20 OU. We followed her over the next four years, and she experienced no recurrence.
1. Rao PK, Rao NA. Vogt-Koyanagi-Harada Disease. In: Ryan SJ (ed.). Retina: Vol. II––Medical Retina, 4th ed. St. Louis: Mosby; 2006:1827-37.
2. Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995 Jan-Feb;39(4):265-92.
3. Arellanes Garcia L, Bautista N, Mora P, et al. HLA-DR is strongly associated with Vogt-Koyanagi-Harada Disease in Mexican Mestizo patients. Ocul Immunol Inflamm. 1998 Jun;6(2):93-100.
Answers
1. d
2. a
3. c
4. d
