 A 20-year-old Hispanic male presented with a chief complaint of slow, painless, progressive vision loss at distance and near. The patient reported that he had difficulty with his vision since he was 10 years of age. His only other symptom was photophobia. His medical and family ocular histories were unremarkable.
A 20-year-old Hispanic male presented with a chief complaint of slow, painless, progressive vision loss at distance and near. The patient reported that he had difficulty with his vision since he was 10 years of age. His only other symptom was photophobia. His medical and family ocular histories were unremarkable.
On examination, his best-corrected visual acuity measured 20/200 O.U. at distance and near. Confrontation visual fields were full to finger counting O.U. His pupils were equally round and reactive to light, with no afferent defect. Extraocular motility testing was normal. His anterior segment examination was unremarkable.
The dilated fundus examination revealed clear vitreous and relatively large optic nerves with moderate-sized cups O.U. The retinal vessels were of normal caliber, and the maculae appeared healthy with a foveal light reflex O.U. We took fundus photographs (figures 1 and 2) and performed a spectral-domain optical coherence tomography (SD-OCT) scan (figures 3 and 4).
Take the Retina Quiz
1. What simple, in-office test would provide the most useful information about our patient?
a. Applanation tonometry.
b. Amsler grid.
c. Color vision testing.
d. Manifest refraction.
2. What additional testing is necessary to help confirm the diagnosis?
a. Electroretinogram (ERG).
b. Electrooculography (EOG).
c. Fluorescein angiography (FA).
d. Visual fields.
3. How would you interpret the SD-OCT?
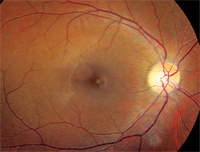
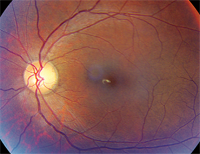
1, 2. Fundus photographs of our patient (O.D. left, O.S. right). What clinical finding do you notice?
a. Normal.
b. Abnormally thick choroid.
c. Loss of the photoreceptor integrity line (PIL).
d. Occult choroidal neovascularization (CNV).
4. What is the most likely diagnosis?
a. Cone dystrophy.
b. Stargardt’s macular dystrophy (SMD).
c. Malingering.
d. Functional vision loss.
For answers, see below.
Discussion
The dilated fundus exam of our patient showed essentially normal optic nerves and what appeared to be healthy maculae. There was no evident macular edema or any other overt macular pathology. In fact, a foveal light reflex was present in each eye.
So, what was wrong with our patient? Did he have some form of functional vision loss, or was our patient a malingerer who was seeking some financial gain? Our answer was embedded in the case history.
One of his primary complaints was photosensitivity. More specifically, he noted that, even on a cloudy day, he was bothered by light. In addition, he believed that he didn’t see as well in normal lighting conditions and that his vision was actually better at night or when the lighting was dimmer. The final clue (which I intentionally omitted from the patient’s history above) was color blindness. In fact, the patient missed all 15 plates O.U. on Ishihara color vision testing.
At this point, all the clues suggested that the patient had a cone dystrophy––although SMD was still a potential differential diagnosis. However, FA testing did not show the classic, quiet choroid that is associated with SMD.
Cone dystrophy is considered to be an acquired disorder that affects the cone photoreceptors. It tends to be progressive in nature and is acquired later in life––unlike other congenital photoreceptor conditions, such as achromatopsia, in which the patient experiences poor vision from birth; nystagmus, an aversion to light (referred to as hemeralopia); and varying degrees of color vision loss.1,2
There are several hereditary patterns of acquired cone dystrophy––all of which result in early loss of color vision as well as a progressive decline in visual acuity (to the level of 20/200 to 20/400).1
In most cases, vision loss begins during the teenage years; however, initial symptoms may present even as late as the seventh decade of life. Interestingly, our patient was first seen at age 15, and at that time his best-corrected acuity was 20/60 O.U. Over the ensuing five years, his vision slowly dropped to the 20/200 level.
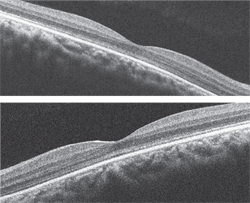
3, 4. The SD-OCT scan of both maculae (O.D. top, O.S. bottom).
The primary difficulty in making the diagnosis is that, in many instances, the retinal exam can be completely normal. Bull’s-eye maculopathy and temporal disc pallor has been reported.1,2 In our patient, the retinal exam appeared normal––although you could almost convince yourself that there may had been temporal optic nerve pallor.
The wide variety of clinical presentations illustrates why electrophysiology is so important in confirming the diagnosis. We performed ERG on our patient, which revealed significantly reduced and prolonged cone response and mildly reduced rod response that was consistent with an acquired cone dystrophy.
The SD-OCT scan is quite interesting. At first glance, it appears normal. However, on careful inspection, you can see that the PIL is absent in the fovea, which suggests that a process is affecting the photoreceptors. The PIL can be seen as a highly reflective line that is located just above the retinal pigment epithelium. It is present outside the macula; but, as you follow it temporal to nasal through the macula, you can see that it disappears on each side.
There are a number of hereditary patterns for cone dystrophy, including autosomal dominant, autosomal recessive and X-linked. Some patients with the X-linked form may exhibit a golden, tapetal-like sheen that disappears on dark adaptation. But, because most cone dystrophies occur sporadically, many researchers believe that the majority of cases are autosomal recessive.1 Our patient’s family history was negative for cone dystrophy, so it is likely that his condition was indeed autosomal recessive in nature.
We referred our patient to the low vision service for evaluation and educated him about helpful programs that were offered by Florida’s Division for Blind Services. Additionally, we completed the necessary forms that declared him legally blind, which will grant him access to special assistance programs.
1. Sunness JS, Carr RE. Retinal Degenerations and Dystrophies. In: Ryan SJ. Retina, vol. I: Medical retina. 4th edition. St. Louis: Mosby; 2006:509-18.
2. Michaelides M, Holder GE, More AT. Inherited Retinal Dystrophies. In: Pediatric Ophthalmology and Strabismus. Taylor D, Greig S (eds). Section 4: Systematic Pediatric Ophthalmology, Chapter 52. 3rd edition. London: Elsevier Saunders; 2005:531-57.
Answers to the Quiz
1. c
2. a
3. c
4. a
