Annual Diagnostic Skills and Techniques IssueFollow the links below to read other articles from our update on diagnostic skills and techniques: 10 Tips and Helpful Hints for Neuro-ophthalmic Disease |
Complex features of retinal and chorioretinal diseases can turn the diagnostic process into a challenge. That’s why learning to identify the nuances that distinguish one disease from another is so critical. In this article, you’ll find five puzzling cases, each accompanied by a self-test designed to build your retinal disease diagnostic chops. (For answers, see key below.)
Case 1: Float On
A 34-year-old black female presented to the clinic for new-onset blurry vision and increased floaters in the right eye. She reported that she was in good medical health and was not on any systemic medications. She was nearsighted and remembered doctors’ reports from prior eye examinations noting retinal scars in both eyes. She noted that this had not changed for a number of years.
The patient had an airbag-related head injury as a result of a motor vehicle accident (MVA) six months prior to this examination. Entering visual acuities were 20/25 OD, 20/25 OS. Chair skills were normal with full to finger counting in both eyes, she demonstrated full extraocular motilities in both eyes, and her pupils were equally reactive to light with no afferent pupillary reaction.
Intraocular pressures (IOPs) were 12mm Hg OD and 10mm Hg OS. The patient’s anterior segment examination was normal, while an evaluation of her posterior segment did reveal abnormal findings (Figures 1 and 2).
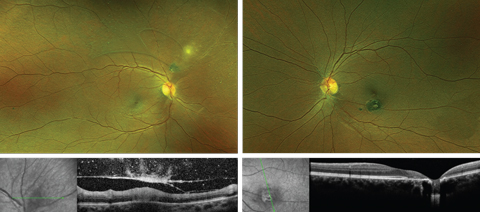 |
| Figs. 1 & 2. Fundus photo and OCT of the right eye (left) and left eye (right). Click images to enlarge. |
1. Which of the following could best explain the new-onset complaint of increased floaters in the right eye?
a. She has had an acute posterior vitreous detachment.
b. The floaters represent normal age-related vitreous condensation.
c. She has developed a rhegmatogenous retinal detachment.
d. The floaters arose from vitritis associated with the pre-existing retinal findings.
2. What is the most likely description of the creamy white-colored lesions superonasal to the optic nerve in the right eye?
a. Superficial retinitis.
b. Cotton wool patch.
c. Coalesced drusen.
d. Retinal exudation.
3. What is the most likely cause of the funduscopic findings?
a. Retinal vascular disease.
b. Infectious disease.
c. Genetic condition.
d. Blunt force trauma.
4. Which of the following is the most likely cause of this symptomatic event?
a. The recent history of MVA.
b. Natural aging process.
c. Reactivation of a preexisting condition.
d. No associated cause.
5. Which of the following laboratory tests would be most useful in confirming the patient’s diagnosis?
a. Angiotensin-converting enzyme.
b. Antinuclear antibodies.
c. Toxoplasmosis serology.
d. Histoplasmosis serology.
Table 1. Serologic Testing and Diagnosis | ||
| IgM | IgG | Interpretation |
| Negative | Positive | Past infection |
| Negative | Negative | No infection; early infection |
| Positive | Negative | Early infection |
| Positive | Positive | Current infection; reactivation of chronic infection |
Case 1 Discussion
The patient has a reactivation of ocular toxoplasmosis, the most common infection of the posterior segment.1 In its active phase, ocular toxoplasmosis causes a superficial retinitis and associated vitritis.2 This typically manifests as a fuzzy or cloudy white overlay on the infected retina, commonly recognized as “headlights in the fog.”2 Retinal vasculitis may also be present.2
When the superficial retinitis eventually advances into deeper tissue, it causes chorioretinal scarring.2 New areas of infected retina often flare near old chorioretinal scars, as in this patient, and are referred to as satellite lesions.2
This condition is caused by the parasitic protozoan Toxoplasma gondii.1,2 Toxoplasmosis can be acquired at any stage of life or contracted congenitally.2 Transmission to humans can occur through undercooked meat, compromised water sources in endemic areas, ingesting dirt or through exposure to cat feces.2 Congenital cases occur via transplacental transmission from an infected mother to the unborn fetus.2
Typically, ocular toxoplasmosis will be diagnosed based on clinical findings alone, but serologic testing is performed to confirm the first-time diagnosis. IgG and IgM antibodies for toxoplasmosis can be ordered in cases with atypical presentations, while IgA can be helpful in determining congenital cases.3 IgG antibodies appear within one to two weeks of infection and reach their peak one to two months post-infection.3
While antibody loads will decrease, they tend to persist at low levels throughout life. IgM antibodies appear within the first week of infection and rapidly increase. Afterwards, IgM decreases to a low level and will disappear after several months (Table 1).3
Case 2: Smells Like Teen Spirit
A 14-year-old Caucasian female presented for an evaluation of retinal holes in both eyes. She was in good health and although her ocular history was remarkable for myopia, her medical history was unremarkable. Her mother added that there was no pertinent family history. Her visual acuities were 20/20 OD, OS. She had normal chair skill testing with full confrontation visual fields, full extraocular motilities and pupils that were equally round and reactive to light with no afferent pupillary defect.
 |  |
| Fig. 3. Retinal imaging of right eye. | Fig. 4. Retinal imaging of left eye. |
The patient had normal anterior segment findings. IOPs were 15mm Hg OD, OS. Retinal findings are available (Figures 3 and 4).
1. The retinal lesions represent which finding?
a. Retinal holes.
b. Hypertrophic retinal pigment epithelium (RPE).
c. Choroidal nevi.
d. Choroidal metastasis.
2. What special ocular imaging/testing would be most helpful in the diagnosis?
a. Visual field.
b. Optical coherence tomographyimaging (OCT) over lesions.
c. OCT angiography.
d. Scleral depression.
3. What specific history should be further questioned in this case?
a. Family history of melanoma.
b. Family history of retinal detachment.
c. Family history of colorectal cancer
d. History of autoimmune disease.
4. What is the proper management of the retinal lesions?
a. Retinal laser.
b. Intravitreal anti-vascular endothelial growth factor (anti-VEGF) agents.
c. Intravitreal steroids.
d. Observation.
5. What referral is necessary?
a. Neurologist.
b. Dermatologist.
c. Gastroenterologist.
d. Rheumatologist.
Case 2 Discussion
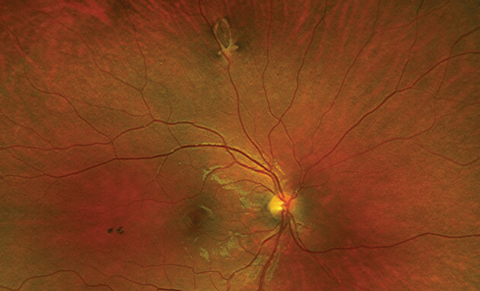
The bilateral lesions noted on fundus examination are the result of hypertrophy of the RPE. In the past, these lesions have been inaccurately defined as congenital hypertrophy of the retinal pigment epithelium (CHRPE). As these lesions are acquired, they are best referred to as RPE hamartomas associated with familial adenomatous polyposis (FAP), abbreviated as RPEH-FAP. 4
FAP is an autosomal dominantly inherited condition in which patients develop multiple polyps in the colon by their teenage years. Left untreated, these patients have almost 100% chance of developing gastrointestinal malignancy.4 When FAP combines with non-GI manifestations such as osteomas, dermoid tumors, cutaneous cysts and other neoplasms, it is known as Gardner syndrome.
 |
| Fig. 5. The top images show typical CHRPE lesion with associated typical OCT findings: thickening and hyperreflectance of the RPE, atrophy of the outer retina with sinking or caving of the inner retina and reduced reflectance of the choroid. The bottom images show typical RPEH-FAP lesions, as well as the variable OCT presentations. Click images to enlarge. |
It is important to recognize the features that distinguish CHRPE lesions from RPEH-FAP. CHRPE, which are mostly benign and have no systemic association, present as unilateral, solitary round lesions with variable pigmentation (Figure 5). They are flat without choroidal involvement or overlying serous detachments. On OCT, a thickening of the RPE will be observed. In areas of lacunae within the CHRPE, thinning of the RPE may exist. Atrophy of the outer retina with excavation of the inner retina over the lesion may also occur (Figure 5).5
RPEH-FAP lesions, on the other hand, are bilateral, multiple, oval or pisciform-shaped and have hypopigmented tails that point posteriorly. On OCT, these lesions may demonstrate choroidal elevation, excavation or both and have overlying shallow serous detachments (Figure 5).6 While these RPE lesions themselves do not require treatment, clinicians must refer patients to a gastrointestinal specialist to examine for colon polyps. Early detection and intervention could prevent cancerous formation.4
Following the fundus examination, we questioned the patient’s accompanying family members about the possibility of a family history of colorectal disease and cancer. The mother reported that both the patient’s father and maternal grandmother have had a history of colon cancer, for which the patient’s father was currently under treatment. The patient was referred to a pediatric gastroenterologist for further management.
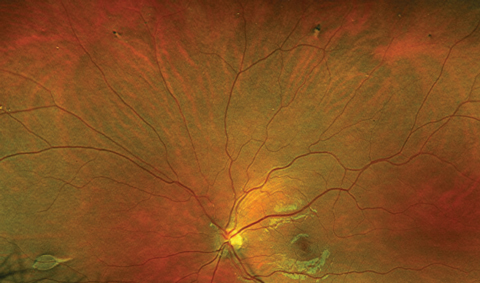
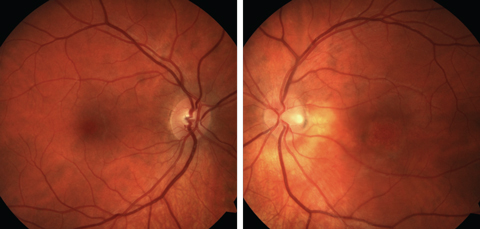 |
| Fig. 6. Fundus photographs of the right and left eyes. Click images to enlarge. |
Case 3: Even Flow
A 29-year-old white male was referred for retinal detachment in his left eye. The patient complained of gradual vision loss in his left eye over the last two months. Medical, past ocular and family histories were all unremarkable. The patient’s best-corrected visual acuities were 20/20 OD, 20/200 OS. Chair skills, confrontation visual fields and extraocular motilities were all normal. His pupils were equally round and reactive to light with no afferent pupillary defect. IOPs were 15mm Hg OD, 18mm Hg OS. Anterior segment findings were normal. Posterior segment findings and OCT findings are depicted (Figures 6 and 7).
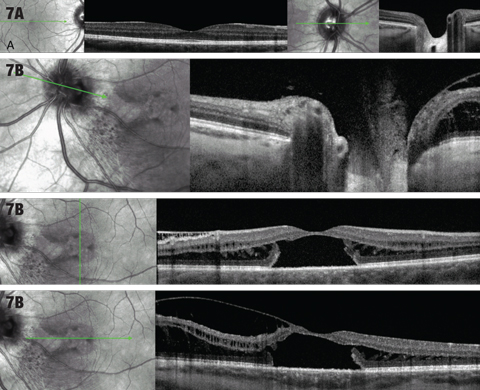 |
| Fig. 7. (A) OCT cross sections through macula and optic nerve of right eye. (B) OCT cross sections through macula and optic nerve of the left eye. Click images to enlarge. |
1. What description is consistent with the macular OCT of the left eye?
a. Choroidal elevation.
b. Cystoid macular edema.
c. Intraretinal fluid and localized serous retinal detachment.
d. Macular hole with vitreomacular traction.
2. The patient’s optic nerve appearance is consistent with which of the following?
a. Glaucomatous cupping.
b. Optic disc pit.
c. Temporal disc pallor.
d. Optic nerve coloboma.
3. Which of the following is the most likely cause of patient’s vision loss?
a. Rhegmatogenous retinal detachment.
b. Choroidal neovascular membrane.
c. Serous retinal detachment.
d. Cystoid macular edema.
4. Which of the following is the most correct diagnosis here?
a. Central serous chorioretinopathy.
b. Choroidal neovascular membrane.
c. Vitreomacular traction syndrome.
d. Optic nerve pit associated maculopathy.
5. Which of the following is the most appropriate treatment?
a. Observation.
b. Topical nonsteroidal anti-inflammatory eye drops.
c. Intravitreal anti-VEGF injection.
d. Surgical vitrectomy.
Case 3 Discussion
This patient has optic disc pit maculopathy. Optic nerve cavitation anomalies exist as a spectrum of conditions that include optic disc pits, morning glory syndrome and optic nerve colobomas.7 With no signs of gender predilection, the prevalence of optic disc pit has been estimated to occur in one out of 10,000 people, and bilateral involvement is found in 10% to 15% of these cases.8
These anomalous disc formations may result in visual field defects, but can also lead to maculopathy when fluid accumulates within the layers of the retina (intraretinal fluid) or in the subretinal space (subretinal fluid or serous retinal detachment). The source of this fluid is still up for debate.9
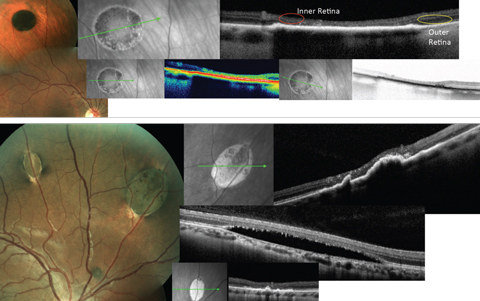
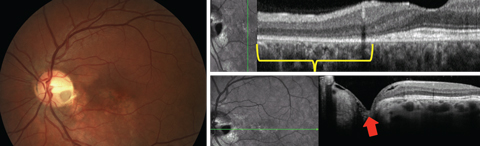 |
| Fig. 8. This patient has an optic disc pit without intraretinal or subretinal fluid. Outer retinal disruption (section in yellow brackets) exists indicative of possible serous detachment in the past. Cavitation in the area of the pit is shown with the red arrow. |
Central vision may be affected by the presence of this subretinal and intraretinal fluids. While the former is often not connected to the disc, the latter typically is.7 OCT imaging of the macula, papillomacular bundle and the optic nerve is helpful in demonstrating the macular changes as well as possible connection of fluid to the optic nerve. It may also reveal anomalous nerve cavitations (Figure 8).
It is important to differentiate optic disc pits from cupping in glaucoma and to monitor these patients for maculopathy. In those with maculopathy, it is necessary to differentiate macular fluid from other causes of retinal edema, such as diabetic macular edema and central serous retinopathy. Patients with optic disc pit that do not show intraretinal fluid or serous retinal detachment do not require treatment. OCT is quite effective in detecting the presence of serous detachments and macular involvement.10
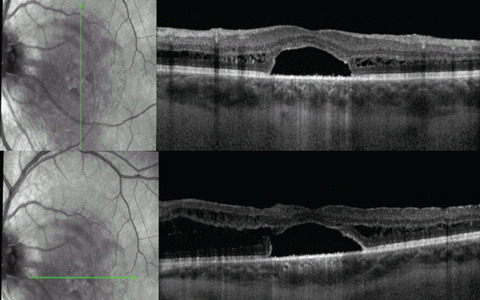 |
| Fig. 9. Macular OCT one month post-op. |
As progressive maculopathy can result in vision loss, surgical intervention may be advised.11,12 This patient underwent pars plana vitrectomy with subthreshold laser temporal to the disc, plus gas injection. At one month post-op, the patient had objective visual improvement as well as structural improvement demonstrated on OCT (Figure 9).
Case 4: Bring Me to Life
A 30-year-old Caucasian female presented to the clinic with recent-onset vision loss and sparkling lights in her left eye for two weeks. She reported taking Prozac (fluoxetine, Eli Lilly) for depression, but had no additional medical conditions. She’s worn lenses for myopic correction since age 13. Her best-corrected entering acuities were 20/20 OD and 20/60 OS. She had full confrontation visual fields and full extraocular motilities. Her pupils were normal with no afferent pupillary defect. IOPs measured 15mm Hg OD and 15mm Hg OS.
Anterior segment findings were normal OU. The posterior segment was normal in the right eye, while OS findings from fundus photography, fluorescein angiography (FA), indocyanine green angiography (ICGA), fundus autofluorescence (FAF) and OCT were suspicious (Figure 10).
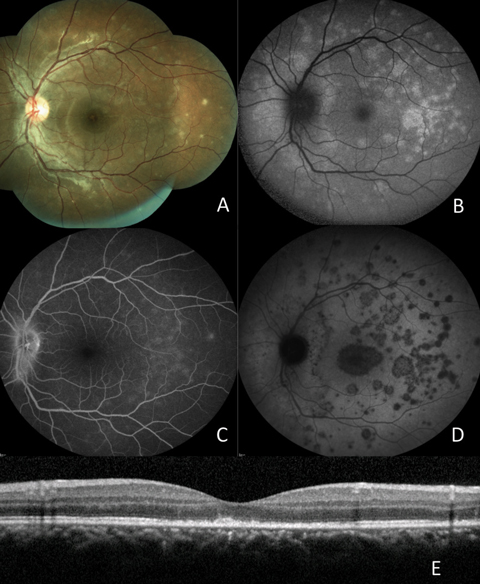 |
| Fig. 10. (A) Fundus photograph; (B) FAF; (C) late FA; (D) late ICGA; (E) OCT. Click images to enlarge. |
1. The white lesions seen on fundus examination represent alterations to which retinal layer?
a. Nerve fiber layer.
b. Inner plexiform layer.
c. Outer nuclear layer.
d. RPE.
2. Which of the following factors has not been reported to be associated with this patient’s diagnosis?
a. Age.
b. Sex.
c. Refractive error.
d. Medication.
3. All of the following can be associated with this condition except:
a. Optic nerve hyperemia.
b. Vitreous cells.
c. Bilateral involvement.
d. Serous retinal detachment.
4. Which of the following are typical visual field findings in this diagnosis?
a. No reduction in visual field.
b. Enlarged blind spot; temporal and paracentral scotomas.
c. Generalized depression.
d. Arcuate defects.
5. What is the most likely etiology of the patient’s condition?
a. Infectious.
b. Degenerative.
c. Autoimmune.
d. Metastatic.
Case 4 Discussion
This patient was diagnosed with multiple evanescent white dot syndrome (MEWDS). It is typically a unilateral condition, but bilateral cases have been reported.13 MEWDS most commonly affects young, white, myopic females. Patients have frequently reported symptoms of paracentral and temporal scotomas, blurred vision and shimmering photopsias.13
MEWDS is an autoimmune inflammatory response that mainly affects the RPE and photoreceptors. Ocular examination reveals multiple white lesions in the fundus. Optic nerve hyperemia may also be present.13 Anterior segment is typically normal, but anterior vitreous cells may be present.13 FA reveals punctate areas of hyperfluorescence, often in a wreathlike pattern and shows hyperfluorescence of the optic nerve in late stages. ICGA, however, reveals early hypofluorescent lesions that remain hypofluorescent in late stages. Lesions seen with ICGA may be more numerous than those seen on examination and FA.14,15
The white spots on a fundus examination appear as hyperfluorescent lesions on FAF. The FAF may also reveal more extensive involvement not seen on fundus examination alone. OCT findings reveal alteration to the RPE and photoreceptors, often with disruption in the photoreceptor integrity line.14,15
MEWDS is diagnosed based on clinical presentation and diagnostic imaging.14,15 While the condition is typically self-limiting, observing these patients for typical progression of the disease and confirming the diagnosis by monitoring gradual self-improvement over time is necessary.14,15
This condition falls under the group of posterior inflammatory “white dot syndromes,” known to masquerade as other ocular inflammatory syndromes.15 Some are more progressive than others, so an accurate diagnosis is essential for proper management. Cases with an abnormal clinical course may require systemic workup to determine specific infectious or inflammatory etiology in need of local or systemic therapy. Patients with white dot syndromes also possess a predilection for choroidal neovascularization, so long-term follow up is imperative.15
Case 5: We Are All Made of Stars
A 30-year-old Caucasian male presented to the clinic for macular evaluation. He reported longstanding vision loss, which worsened progressively throughout his childhood and teenage years. Leading up to the visit, he had not noticed any recent changes to his vision. His medical history was positive for seasonal allergies, for which he used Claritin (loratadine, Merck). The patient reported no family history of systemic or eye disease.
Best-corrected visual acuities were 20/200 OD and 20/150 OS. He had normal chair skill testing with full confrontation visual fields, normal extraocular motilities and pupils that were equally round and reactive to light. His IOPs measured 15mm Hg OD and 16mm Hg OS. Anterior segment findings were also normal.
The patient’s posterior segment findings include fundus photography, FAF and macular OCT findings (Figure 11).
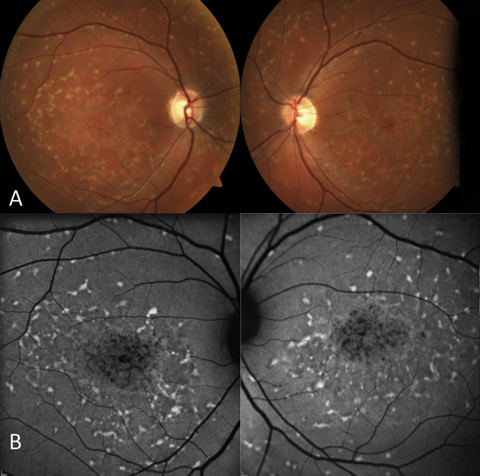 | 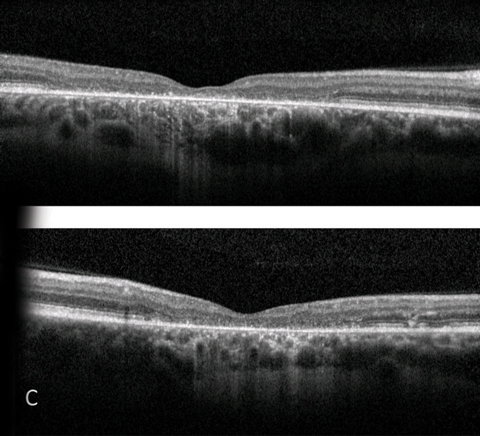 |
| Fig. 11. (A) Fundus photograph; (B) FAF; (C) OCT. | |
1. Hyper-autofluorescent areas on the FAF correspond to:
a. RPE atrophy.
b. Buildup of lipofuscin.
c. Drusen.
d. Pigment epithelial detachments.
2. Hypo-autofluorescent areas on the FAF correspond to:
a. RPE atrophy.
b. Buildup of lipofuscin.
c. Drusen.
d. Pigment epithelial detachments.
3. OCT imaging reveals:
a. Atrophy of vitreoretinal interface and epiretinal membrane formation.
b. Retinal nerve fiber layer atrophy.
c. Retinal ganglion cell atrophy.
d. Atrophy of the outer retina, including RPE and photoreceptors.
4. The hyper-reflectance of the subfoveal choroid on the OCT is caused by:
a. Overlying nerve fiber layer atrophy.
b. The thickening of the choroidal vessels.
c. Atrophy of the overlying RPE.
d. The upward displacement of the choriocapillaris.
5. Abnormal findings are to be expected for each of the following diagnostic tests except:
a. Visual field testing.
b. Ophthalmodynamometry.
c. ERG.
d. Color vision.
Case 5 Discussion
The patient’s diagnosis is Stargardt disease. The most commonly occurring inherited macular dystrophy, it is inherited in an autosomal recessive fashion.16 The gene responsible, ABCA4, codes for a protein located on the photoreceptor’s outer segment discs. This protein is involved in the transport of all-trans-retinal (atRAL) out of the outer segments so it can be properly metabolized by the RPE. Improper transportation results in accumulation of waste material and lipofuscin. In addition to Stargardt disease, the ABCA4 gene is implicated in many other conditions such as cone-rod dystrophies, retinitis pigmentosa and age-related macular degeneration.16
As there are many variations of gene mutation in ABCA4, there are also different phenotypical presentations of Stargardt.16 A hallmark characteristic is yellow-white pisciform-shaped flecks at the level of the RPE. These lesions may be present in addition to foveal atrophy that is classically described as having a “beaten bronze” appearance, or the flecks may present in isolation. In the past, pisciform lesions in the absence of macular atrophy was termed fundus flavimaculatus; recently, most agree they are different phenoytypes of the same disease.16
Patients affected by Stargardt disease typically begin to notice vision loss in their teenage years, falling on the spectrum of mild loss to 20/200 or worse.16 Initial fundus findings may be subtle, making the condition challenging to diagnose early. Patient symptoms may seem exaggerated when compared with fundus examination in early stages of the disease. FAF and OCT imaging, however, may reveal subtle alterations to the RPE and outer retina even before fundus alterations are visible.16 For this reason, these tools are quite useful for early diagnosis. Patients may also develop red-green color defects, visual field defects and may have abnormal ERG findings.16
Answer KeyCase 1: (1) d; (2) a; (3) b; (4) c; (5) c. |
OCT in Stargardt disease can reveal various levels of RPE and photoreceptor atrophy. In this case, there was significant outer retinal atrophy, loss of visibility of the photoreceptor integrity line and external limiting membrane (ELM) throughout a large area of the macula. This directly correlates with vision loss in the patient. In certain stages of the disease, reports note a thickening of the ELM on OCT.17 In addition, flecks may present as subretinal hyper-reflective deposits with various disruption of overlying photoreceptor layers.18
There are also various autofluorescent patterns found across the many different clinical manifestations of Stargardt disease. Flecks are hyper-autofluorescent, while areas of RPE atrophy will be hypo-autofluorescent. Bull’s-eye autofluorescent patterns can also be found.19
Diagnosis of the condition is typically made based on fundus examination, FAF and OCT testing; however, a definitive diagnosis would require genetic testing to confirm the genetic mutation. No current treatment for Stargardt disease exists, but patients should be monitored for development of choroidal neovascular membranes.20
Taking steps to strengthen your diagnostic acumen on a regular basis is critical to your practice. This exercise can provide you with the tools you need to provide sound, thorough care to your patients.
Dr. Rafieteery is a consultative optometrist at the Charles Retina Institute in Germantown, Tenn.
Dr. Haynes is a clinical optometric fellow at the Charles Retina Institute.
1. Kijlstra A, Petersen E. Epidemiology, pathophysiology, and the future of ocular toxoplasmosis. Ocular ImmunolInflamm. 2014;22(2):138-47. |
