Demyelinating disorders represent a heterogenous group of central nervous system (CNS) pathologies characterized by the loss of myelin sheath or the cells that form them. Primary demyelination of the CNS represents a category within a broad spectrum of inflammatory autoimmune disorders that occurs against the backdrop of chronic inflammation and neurodegeneration. Multiple sclerosis (MS) remains the most common primary CNS demyelinating disorder, affecting white and grey matter of the brain, spinal cord and optic nerve.1 Until recently, neuromyelitis optica (NMO) spectrum disorder and myelin oligodendrocyte glycoprotein (MOG) antibody disease represented variants of the disease despite their distinct pathologic and phenotypic expressions.
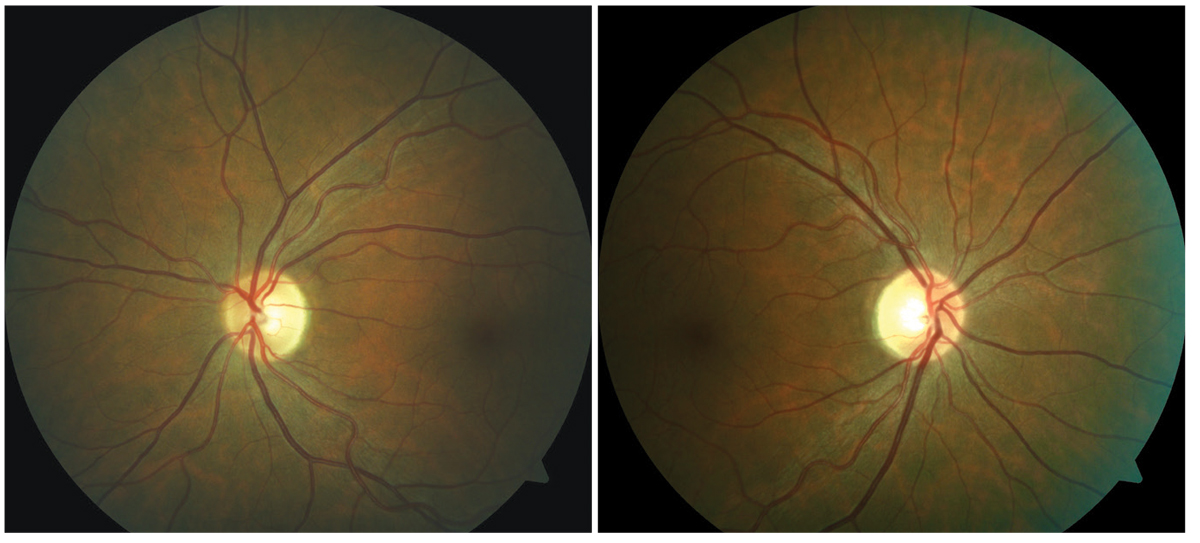 |
|
Asymptomatic bi-temporal optic disc pallor representing progressive demyelination of the optic nerve without a history of optic neuritis. Click image to enlarge. |
The advent of antibody testing for NMO spectrum disorder (AQP4-IgG) and MOG antibody disease (anti-MOG) has enabled clinicians to better differentiate among each disease and more accurately determine the prognosis and response to therapy.
Optic neuritis (ON) is a general term that is used to describe inflammation of the optic nerve that can occur in isolation or as a manifestation of a systemic disease process. Numerous etiologies are responsible for ON and broadly classified as typical or atypical based on clinical, laboratory and imaging findings. The aim of this article is to compare and contrast the immunopathology, clinical presentation, diagnostic criteria, and management of MS, NMO spectrum disorder and MOG antibody disease in the setting of typical and atypical ON. Additionally, systemic causes of atypical ON will be reviewed.
 |
| Click table to enlarge. |
Typical Optic Neuritis
This classification describes an underlying demyelinating process that occurs in isolation or as a clinical manifestation of MS. Performing comprehensive ophthalmologic clinical examination, including neuroimaging and studies and laboratory evaluation, is necessary to differentiate the three primary demyelinating diseases.
In cases of ON with two or more non-contrast enhancing lesions on magnetic resonance imaging (MRI), a diagnosis of clinically isolated syndrome can be made, which is associated with a high risk of developing MS. Suspected cases of acute demyelinating ON should undergo a comprehensive ophthalmologic examination and ancillary testing (Table 1).2
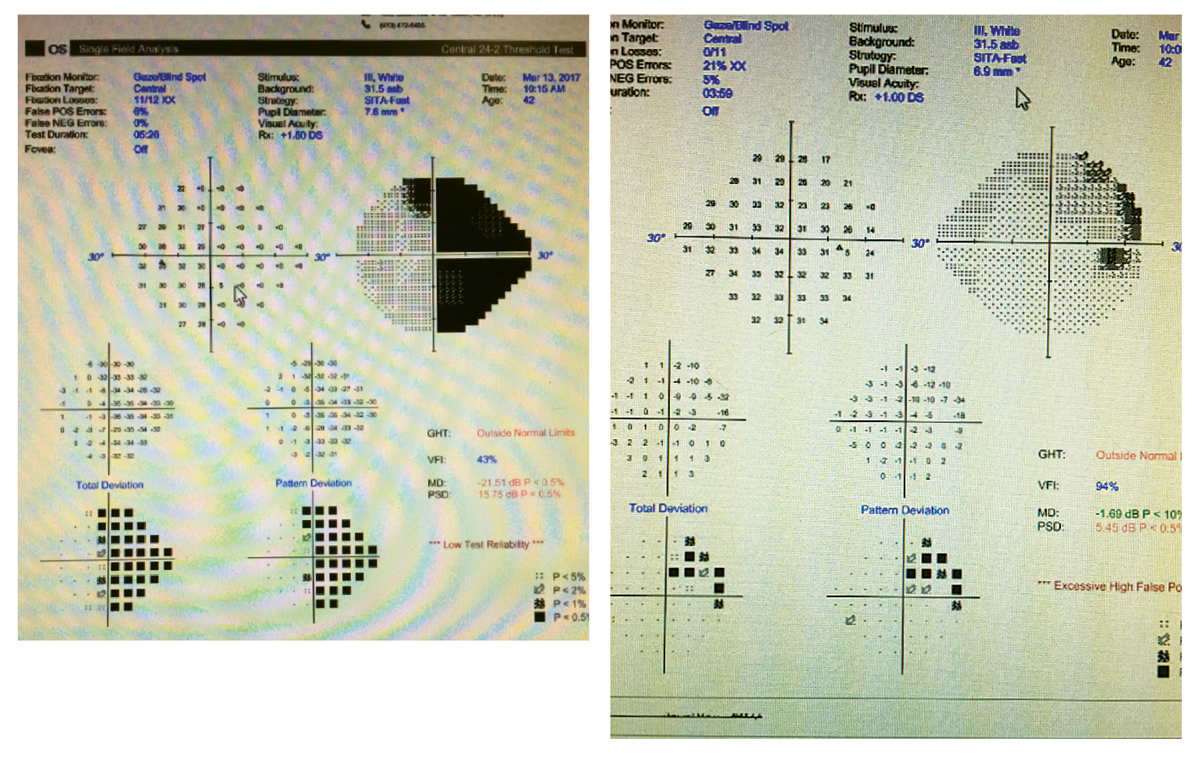 |
|
Partial non-congruous right hemianopia in a patient with relapsing-remitting MS. Click image to enlarge. |
Acute demyelinating ON is the presenting sign of MS in about 20% of patients, and will occur over the course of the disease in another 50%.3 In the general population, the annual incidence of new-onset acute demyelinating optic neuritis has been reported to be 0.5 to 5.1 per 100,000.4 The disease typically affects Caucasians with a female predominance of two to one.2
The most common presentation of typical acute demyelinating ON includes acute unilateral loss of vision accompanied by pain on eye movement, diffuse and central scotoma, and dyschromatopsia.5 Additional complaints include recurring photopsia, exercise or heat-induced vision loss (i.e., Uhthoff phenomenon) and anomalous perception to moving objects (i.e., Pulfrich effect), the latter of which develops as vision improves.5
 |
| Click table to enlarge. |
The degree of vision loss can range from normal visual acuity to no light perception with a decrease in contrast sensitivity. Pain and eye movement that precedes acute central vision loss with a normal or mildly edematous optic disc edema is the most common clinical features observed in MS-related optic neuritis. When present, these key clinical features can be pathognomonic for a demyelinating event.
Following an acute episode of typical acute demyelinating ON, patients will experience near normal improvement in visual acuity, visual field and color vision, with the exception for contrast perception. Typically, the loss of acuity is described as taking place over hours to days with gradual improvement over days to weeks.6 Visual field defects are commonly observed with diffuse central scotomas occurring most frequently.7 Swelling and hyperemia of the optic disc (i.e., papillitis) is observed in only one-third of cases, while the remaining two-thirds appear normal (i.e., retrobulbar ON).3
Neuroimaging is required to determine the visual and neurologic prognosis, and to differentiate among alternative demyelinating diseases. In contrast to acute typical ON, optic nerve pallor is a finding that suggests longstanding, chronic demyelination secondary to MS. In the absence of key features that suggest an isolated demyelinating event or MS-related ON, there is a greater likelihood that the patient has atypical ON.
MRI of the brain with and without contrast is the most important test ordered to determine systemic demyelination as well as to understand the visual and neurological prognosis. Ideally, it is recommended to include contrast-enhanced imaging of the orbits, in order to assess the level of activity, and to discount masquerading diseases such as orbital tumors (e.g., meningioma) and lymphoproliferative lesions (e.g., leukemia and lymphoma). In the setting of acute demyelinating ON, contrast enhancement of the optic nerve on MRI has been observed in 94% of patients and has distinguishing features that differentiate it from NMO spectrum disorder and MOG antibody disease (Table 2).8
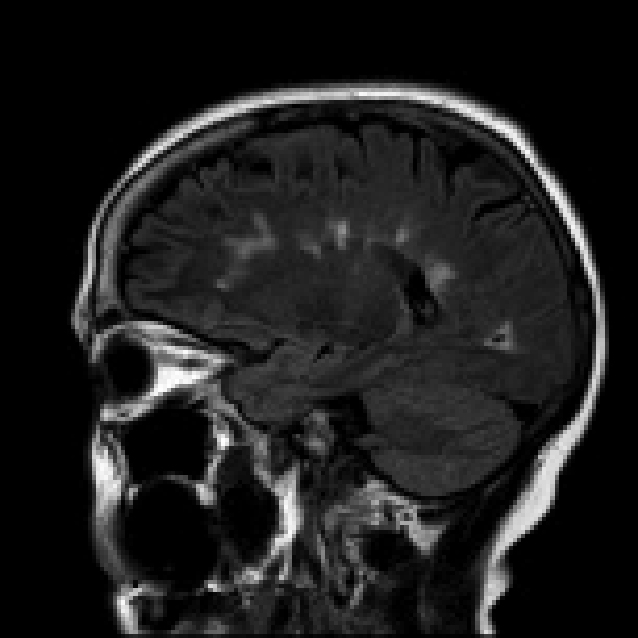 |
|
Demyelinating plaques within the cerebral hemispheres. Click image to enlarge. |
While there is no one single biomarker that can confirm the presence of MS, the revised McDonald criteria provides supportive data that is used to establish the diagnosis and assess the response to therapy (Table 3).9 In the setting of typical ON, abnormal neuroimaging increases the probability of developing MS at 15 years from 25% to 72%. On the basis of the Optic Neuritis Treatment Trial (ONTT), the five and 10-year risk for developing recurrent ON are 28% and 35%, respectively, occurring more frequently in patients with MS.5
Ancillary testing (e.g., cerebrospinal fluid (CSF) analysis and blood work) are not considered standard procedures when the clinical findings are consistent with MS-related ON. Analyzing the CSF has value when MRI findings are not definitive and when the clinical findings are not consistent with MS. Laboratory testing is also not an international standard and is reserved for cases that suggest an alternative etiology. In a large cohort of patients, testing for antinuclear antibodies, syphilis serologies and chest X-ray were found to have no clinical indigence.5
Following a diagnosis of MS-related ON, treatment for MS is aimed at treating the acute attack and reducing the frequency of attacks. For acute attacks, intravitreal (IV) methylprednisolone is the standard treatment.5 It has been well-established that three to five days of treatment with IV methylprednisolone leads to faster improvement in visual recovery but has no effect on the finale outcome in vision.8 Following guidelines from the ONTT, IV methylprednisolone followed by oral prednisone resulted in lower rates of MS within the first two years.5 Plasma exchange can be used if the attacks are severe and the disease is recalcitrant to corticosteroids.8 Long-term immunosuppression is recommended to reduce morbidity and mortality. Treatment options for relapsing and remitting MS include injectable, oral and IV infusion.9
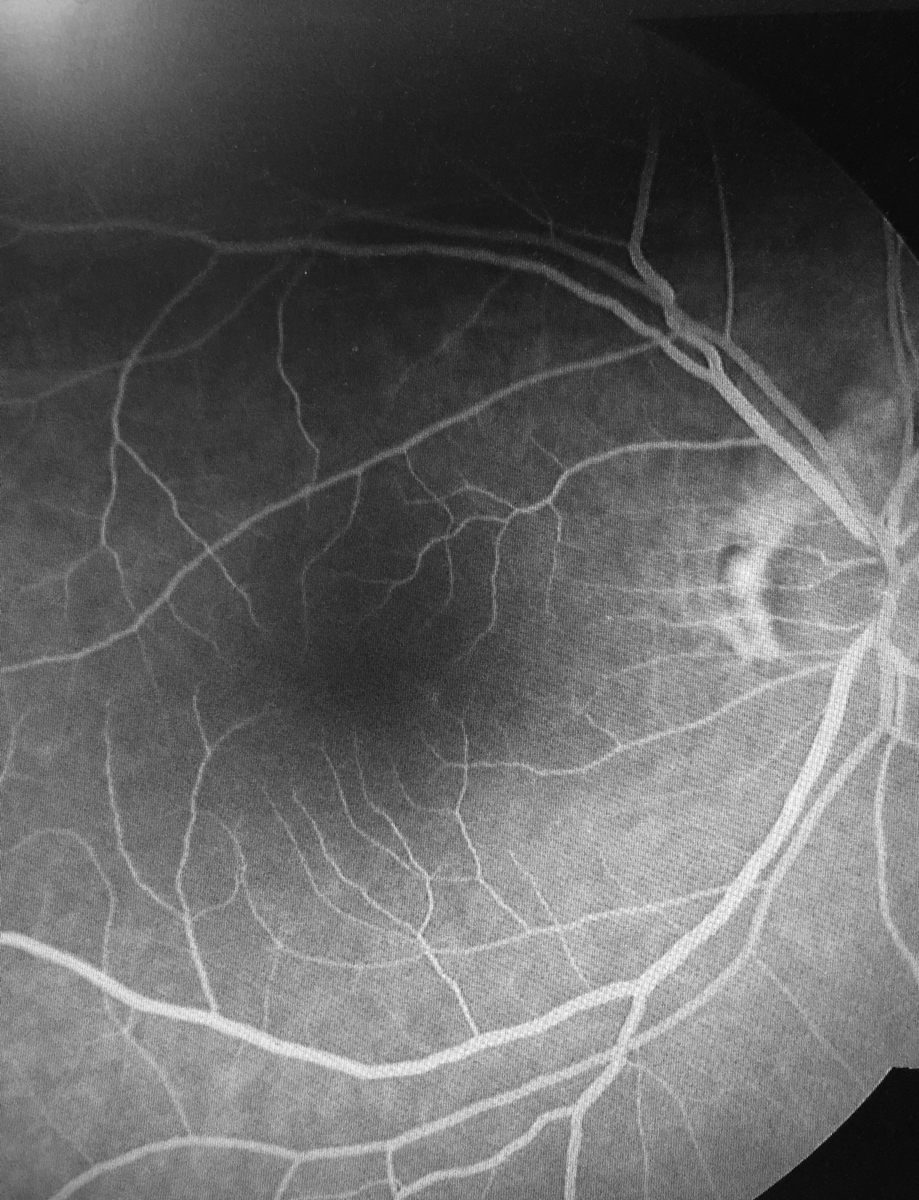 |
|
Normal fluorescein angiography of the right eye in a patient with acute vision loss attributed to acute ON. Click image to enlarge. |
Fingolimod became the first drug indicated for treating relapsing MS; it is a once-daily oral medication that works by blocking lymphocytes from escaping the lymph and traveling into the CNS. Siponimod is also an oral medication taken daily and the first that is indicated for to treat secondary progressive disease by inhibiting both T and B cells. Ocrelizumab is approved for all forms of MS (e.g., relapsing-remitting, primary progressive and secondary progressive). It is given in an IV infusion every six weeks and works by destroying B cells. Ofatumumab is an injection given once a month. It too targets B cells and is approved for relapsing MS.
It is important to understand both the form of the disease as well as the medication in order better assess the visual status and confidently address patient concerns. The short-term goal in disease-modifying agents is to decrease the amount of lesion activity as detected on MRI. After initiating therapy, the goal of the neurologist is to monitor for progression and identify early signs of toxicity.8
Multiple Sclerosis
Primary demyelinating disorders are chronic inflammatory disorders of the CNS characterized by cell-mediated autoimmunity and neurodegeneration of brain, spinal cord and optic nerve.11 Despite the elusive nature of its pathogenesis, there are several factors that have been implicated in MS: infection (e.g., Epstein-Barr virus and human herpes virus-6), low vitamin D, geographic gradient, obesity, smoking and genetic factors.12 With over 200 identified genetic inheritance patterns, HLA-DRB1 represents the most common variant.13
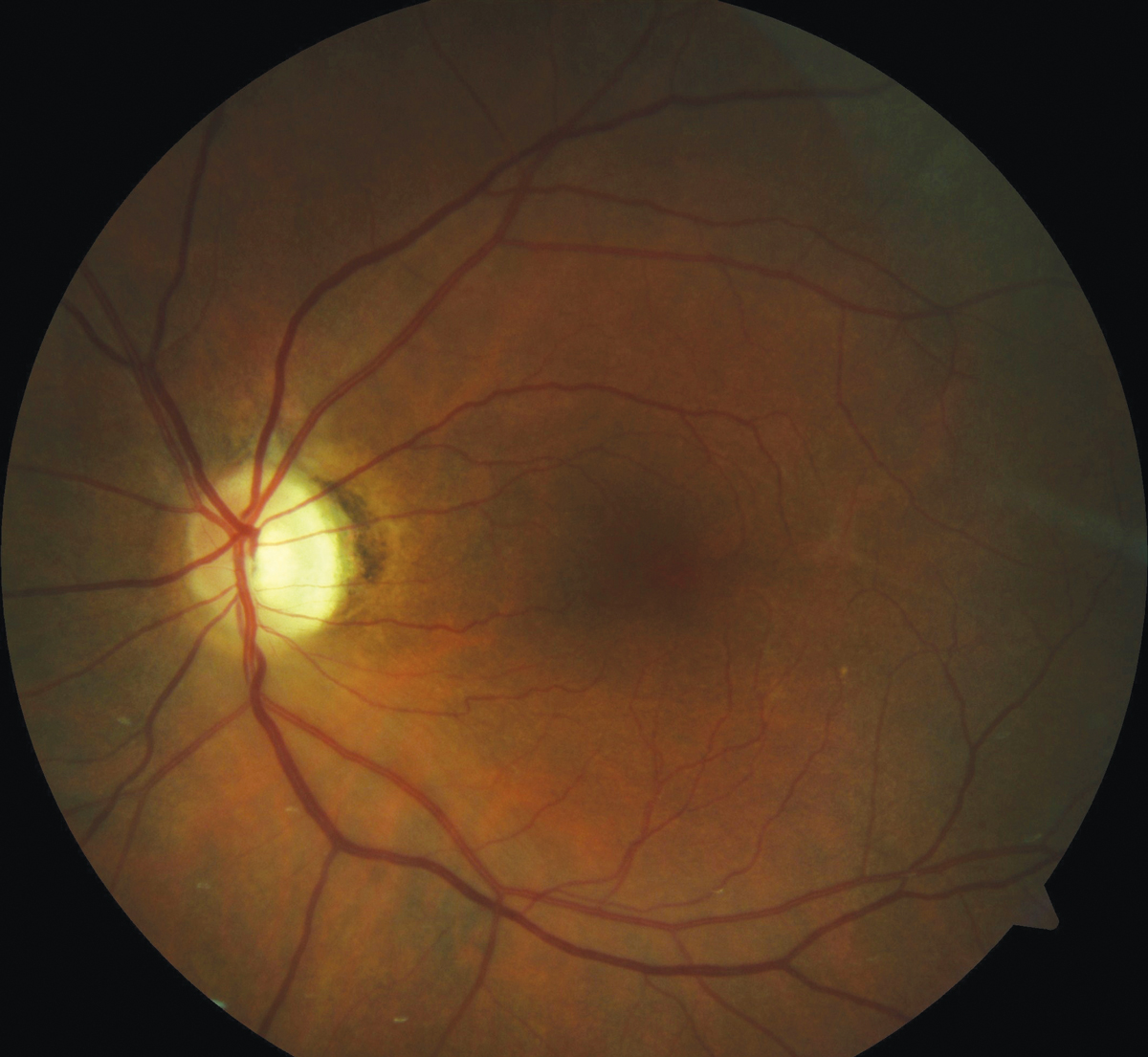 |
|
Optic nerve pallor of the left eye following resolution of retrobulbar ON. Click image to enlarge. |
The annual incidence of MS is 2.1 cases per 100,000 with a prevalence rate of 35.9 per 100,000 worldwide.13 Women are affected more than men with a predominance of two to one, and highest among populations that are more distant to the equator.12 In the CNS, myelin sheaths are formed and maintained by oligodendrocytes. Both the adaptive and innate immune system are responsible for stripping the myelin lamellae and removing myelin fragments.
Demyelination represents the final common pathway of MS. While acute demyelinating lesions can occur anywhere in the CNS, there is a predilection for areas with high venous density as inflammatory cells egress through veins from blood to brain (e.g., Dawson fingers).14,15 Contrary to acute lesions, smoldering demyelinating lesions are often associated with progressive MS.15 These lesions are characterized by loss of peripheral axons that are replaced with iron-containing microglia, hypothesized as a CNS-resident immune response.15,16 Lastly, chronic lesions are characterized by circulating T cells and macrophages surrounding an area that is hypocellular.16,17 Treatment for primary MS is similar to MS-related ON discussed previously.
NMO Spectrum Disorder
First described in the late 19th century as simultaneous acute demyelinating ON and myelitis, NMO is now considered just one of a number of phenotypes that represent NMO spectrum disorder, particularly reflected by prominent perivascular immunoglobulin deposition and complement activation with autoantibodies to aquaporin-4 (AQP4).18,19 Patients with signs and symptoms of NMO spectrum disorder require serum antibody testing via cell-based assay of AQP4-IgG plus contrast-MRI of the brain, orbits and spinal cord.9
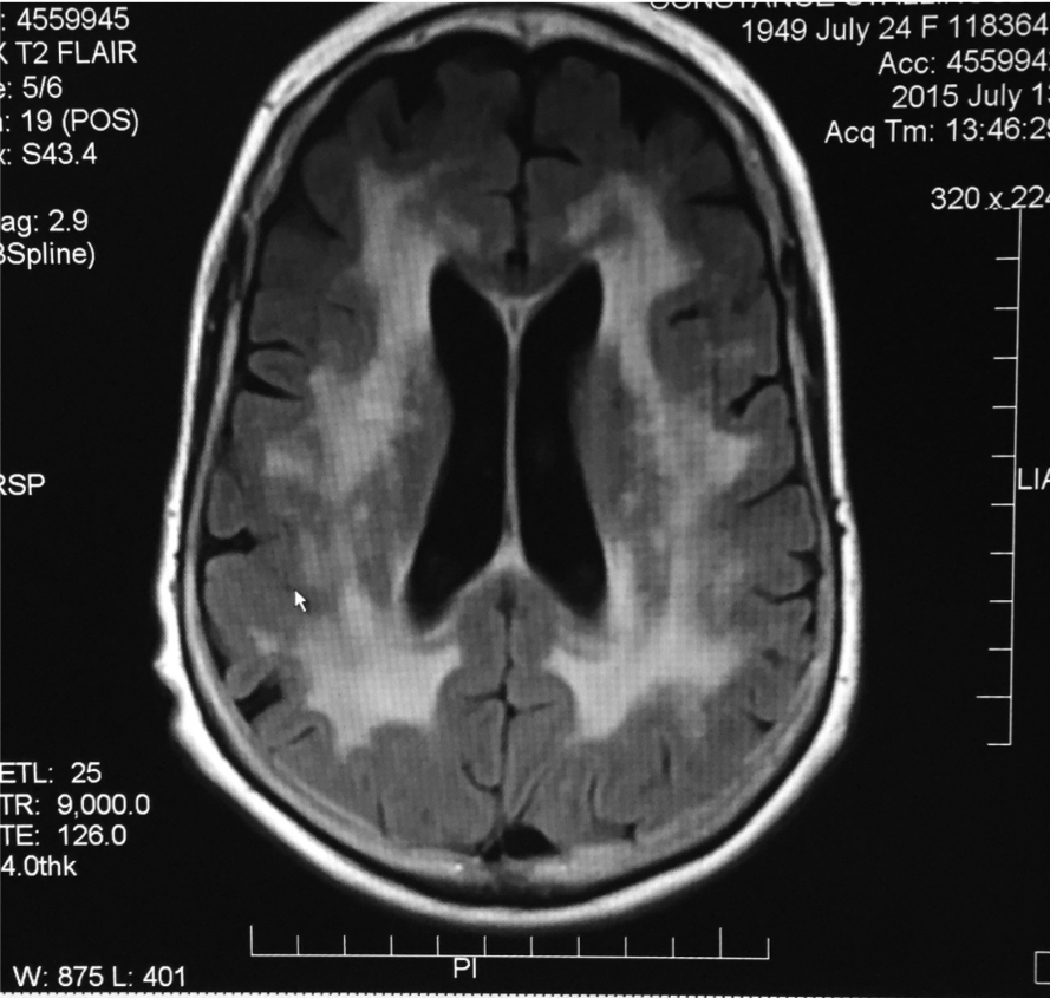 |
|
Axial MRI T2 FLAIR showing confluent white matter lesions representing demyelination from leukoencephalopathy. Click image to enlarge. |
The mean time from the first symptom to relapse is 8.5 months regardless of AQP4-IgG status. However, the presence of AQP-4 antibodies increases the likelihood of severe visual impairment, recurrence of ON, and subsequent development of transverse myelitis; longitudinally extensive transverse myelitis describes inflammation involving a minimum of three contiguous vertebral segments.9,20
NMO spectrum disorders affect women more than men. Unlike MS, these disorders are often found in non-Caucasians with a significantly lower incidence and prevalence rate of 0.053 to 0.40 per 100,000 and 0.52 to 4.4 per 100,000, respectively.20,21 Core clinical features of NMOSD include acute demyelinating ON, myelitis, area postrema and other brainstem syndromes, as well as diencephalic and cerebral sign.22 Systemic symptoms include intractable nausea and vomiting, hearing loss, diplopia, vertigo, facial palsy and sleep disturbances.22
Aside from acute demyelinating ON and myelitis, the diagnostic criteria for NMO spectrum disorder has expanded to include a larger topographic area of CNS involvement and seropositive AQP-4-IgG.23 Clinically, the diagnosis of NMO spectrum disorder is most accurately characterized as NMO spectrum disorder with AQP4-IgG or without AQP4-IgG.23,24 Traditionally, acute attacks are treated with IV methylprednisolone and plasma exchange; however, up to 50% of patients experience a recurrence in symptoms.24 Additional broad-spectrum immunosuppressive agents have also been used but with lower efficacy as compared to newer targeted therapies.
There have been three recently approved monoclonal antibody medications: rituximab for B cell inhibition; eculizumab, a complement inhibitor; inebilizumab, an anti-CD9 agent; and satralizumab, an anti-leukin 6 receptor. All therapeutic agents target specific disease pathways in patients with AQP-4 antibodies and have been shown to be safe and effective for long-term immunotherapy, as traditional MS medications may increase the rate of relapse.24 Specifically, satralizumab is effective for AQP-4 seronegative NMO spectrum disorder, and in patients with AQP-4 seropositive disease, eculizumab was found to have a 94% reduction in the rate of relapse.
 |
| Click table to enlarge. |
 |
| Click table to enlarge. |
Acute Disseminated Encephalomyelitis (ADEM) and MOG Antibody Disease
ADEM is a monophasic demyelinating disorder of the CNS, most prevalent in children, characterized by extensive macrophage involvement.25 CNS involvement usually shows small, non-confluent, demyelinating lesions within the perivascular region. This distinguishing factor differentiates it from large confluent perivenous lesions observed in MS.25 Demyelinating lesions in ADEM can occur throughout the CNS, including the white matter, cortex, thalamus and basal ganglia. ADEM typically affects children and young adults following seasonal changes, infection or vaccination (e.g., Epstein-Barr, human herpes virus-6 and coronavirus).26
MOG is a minor myelin protein that is expressed on the surface of myelin sheaths of the CNS and is targeted by IgG1 antibodies.27 Lesions are typically characterized as large, hazy, bilateral and with vast topographic involvement, including longitudinally extensive transverse myelitis.27 Similar to NMO spectrum disorder, this form of myelitis usually has greater proclivity for the thoracic cord.27
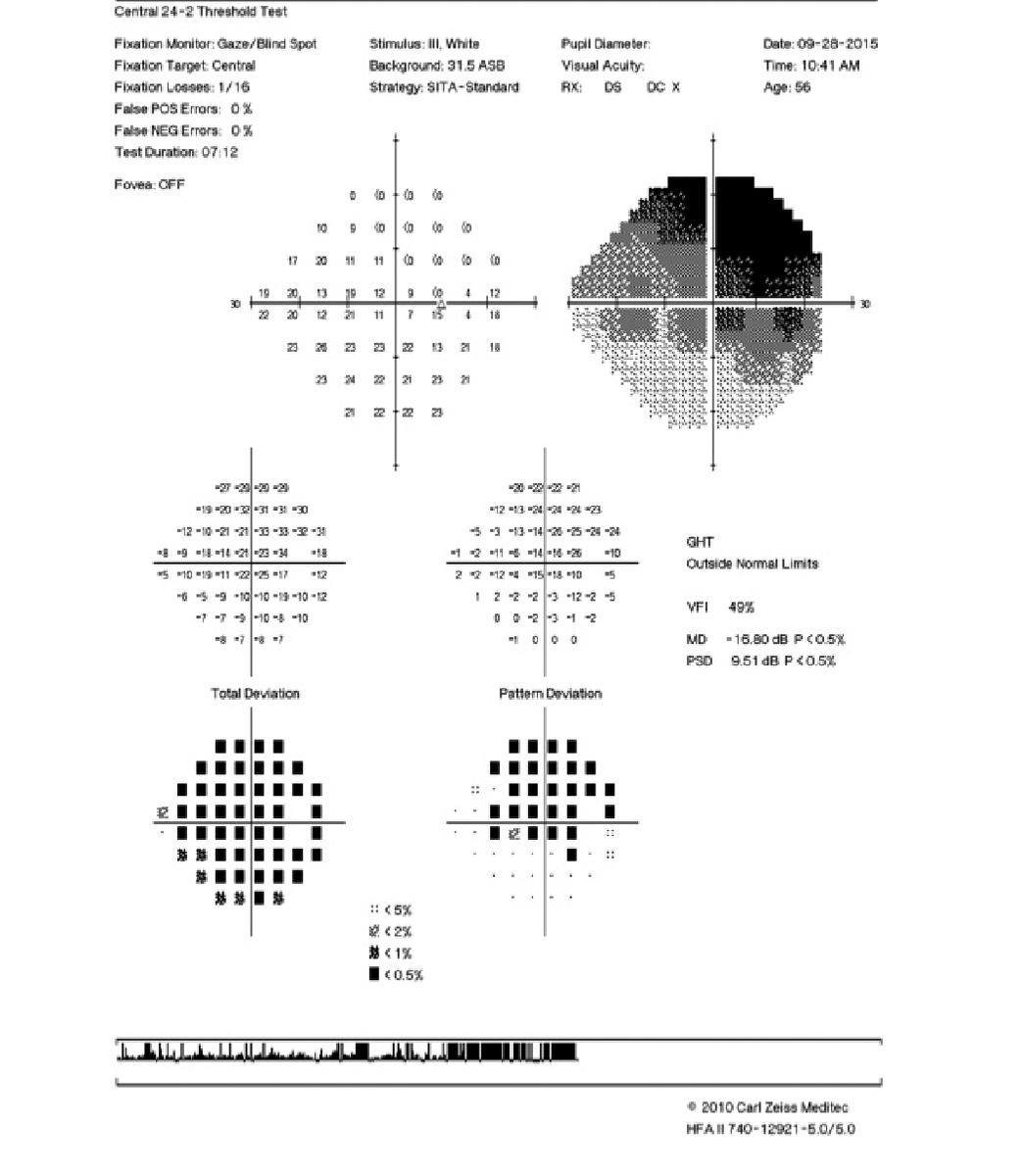 |
|
Incomplete superior visual field scotoma involving fixation. Click image to enlarge. |
In children, up to 60% with MOG antibody disorder have bilateral thalamic involvement and lesions within the cerebellar peduncle.28 Just over half of these patients have short hyperintense lesions within the thoracic cord.28 CSF analysis has been shown to express pleocytosis, elevated protein, and limited oligoclonal bands. Cell based serum assays are recommended for detecting MOG-IgG when clinical features are consistent with a diagnosis of MOG antibody disease.28
Bilateral, simultaneous acute demyelinating ON is the most common initial presenting sign, reported to occur in 51% of MOG antibody disease-related ON patients.29 Pain on eye movement and papillitis are two of the most common clinical features. While loss of vision is typically severe in the acute stage, less than half of patients present with normal brain MRI.30 In contrast, MOG antibody disease testing has been shown to be highly specific and sensitive during an acute attack.29 This highlights the importance of both early detection and timely serologies. As compared with both NMO spectrum disorder-related ON and MS-related ON, clinical and paraclinical findings in MOG antibody disease show both similarities and differences (Table 4).29
Timely diagnosis helps to establish the prognosis and choose the most appropriate treatment, which is to eliminate systemic MOG antibodies and improve the visual outcome. Typically, treatment consists of IV methylprednisolone based on weight. In cases where there is a weak response to IV methylprednisolone or recurrence, either intravenous immunoglobulin or plasma exchange are additional options. The symptoms of recurrent MOG antibody disease-related ON can also be dampened using disease modifying agents. Because poor visual acuity has been reported in 16% of patients, timely and judicious management is necessary to preserve visual function and quality of life.31
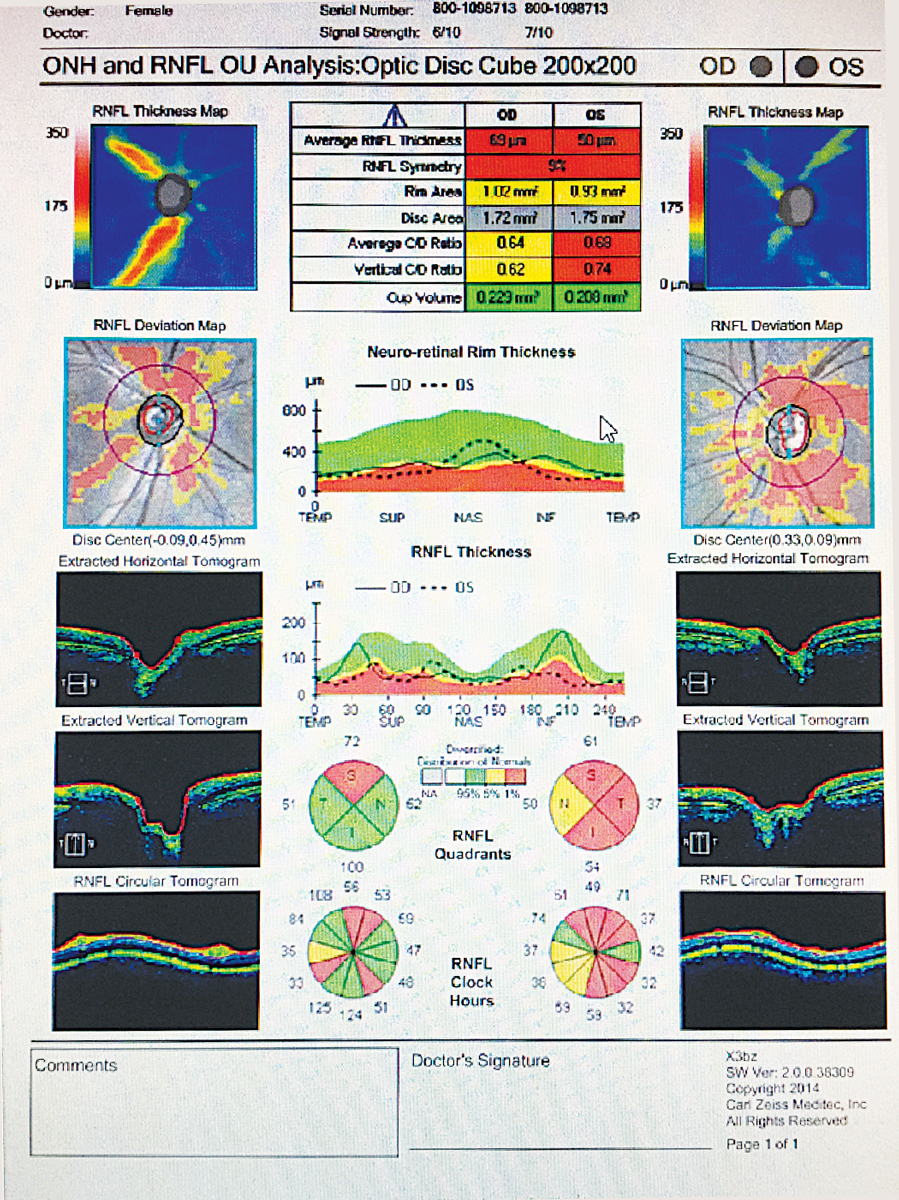 |
|
Bilateral retinal nerve fiber loss OU in a patient with MS without a history of ON. Click image to enlarge. |
Acute treatment for MOG antibody disease is IV methylprednisolone and plasma exchange. Disease modifying agents used for long-term therapy include IV immunoglobulin, rituximab, mycophenolate mofetil, methotrexate and azathioprine. The role of steroids as adjunct treatment with immunosuppressants still remains unanswered.28
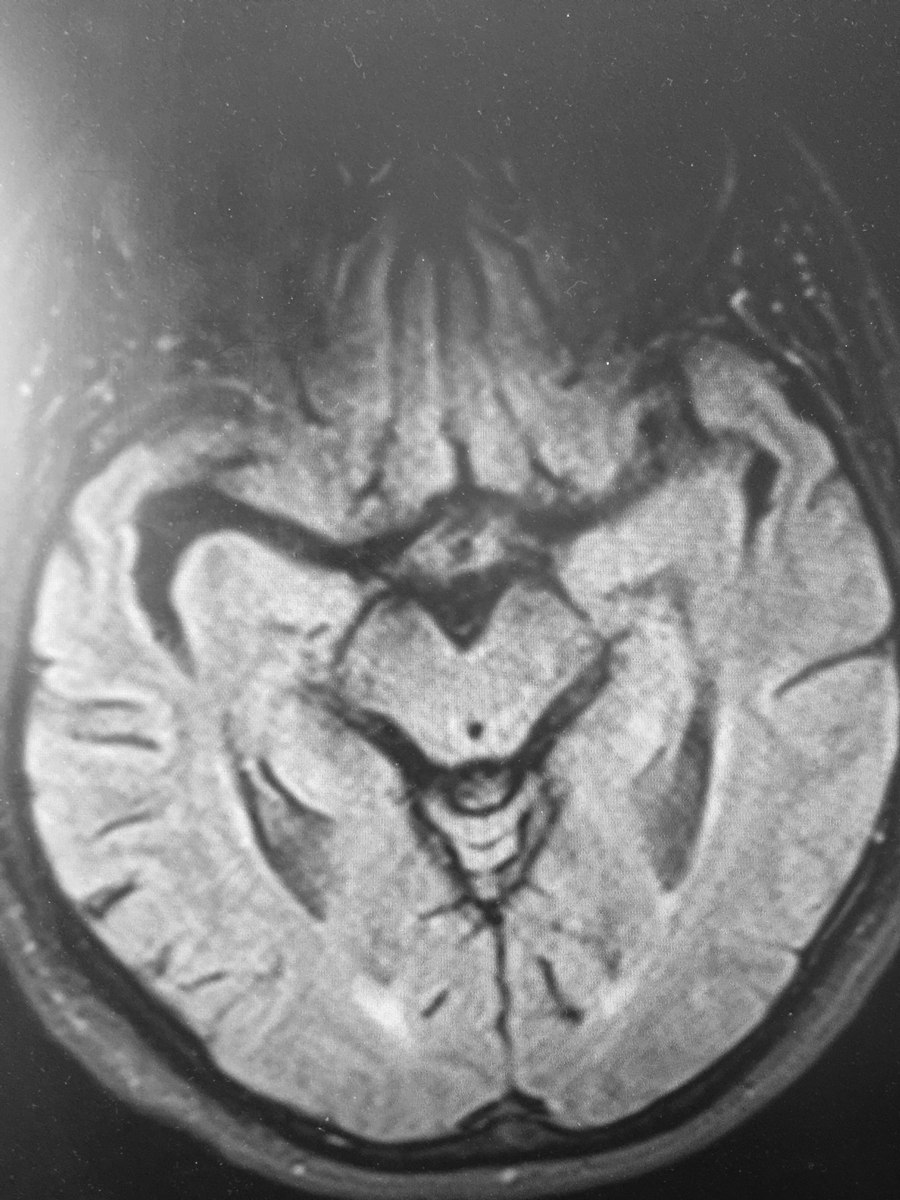 |
|
Multifocal white matter lesions in a patient with relapsing and remitting MS. Click image to enlarge. |
Takeaways
Knowing which clinical features are atypical for ON (e.g., bilateral, poor visual recovery) and which are consistent with MS, NMO spectrum disorder and MOG antibody disease is important for preserving vision and quality of life.
A patient who is suspected of optic neuritis needs comprehensive eye examination, including ancillary testing (i.e., OCT, fundus photography and automated perimetry). In the absence of systemic disease and atypical findings, the patient needs emergent contrast-enhanced MRI of the brain and orbits with and without contrast and fat suppression. Findings that are consistent with MS-related ON should receive IV methylprednisolone followed oral corticosteroids in order to speed visual recovery and reduce the risk of developing MS. Additional therapy is recommended for long-term immunosuppressive therapy. The optometrist should perform serial visual fields, OCT and fundus photography. Comanagement with a neurologist who subspecializes in MS is recommended.
In the presence of a history of systemic disease (e.g., autoimmune disorders or infection), abnormal neuroimaging or ophthalmologic findings consistent with atypical ON, the patient requires additional contrast-enhanced neuroimaging of the spine along with laboratory tests and possible CSF analysis. It is especially important to differentiate among the three primary demyelinating diseases, as they all initially present with ON in absence of other ophthalmologic findings. In these cases, it is recommended to refer or admit the patient to the ER for contrast-enhanced MRI of the brain, orbits and spine, in addition to NMO-IgG and anti-MOG.
Optometrists need to recognize the early signs of demyelinating disease in the eye. Rapid, accurate diagnosis will allow the patient to receive the appropriate treatments and have the best chance at maintaining vision and a high quality of life.
Dr. DelGiodice previously practiced at Associated Eye Physicians in Clifton, NJ, and recently accepted a position as a medical science liaison for Sun Pharmaceuticals. His industry role had no bearing on the content of this article. Dr. DelGiodice is also a Fellow of the American Academy of Optometry.
1. Hersh CM, Fox RJ. Multiple sclerosis. Cleveland Clinic Center for Continuing Education. Published April 2018. www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/neurology/multiple_sclerosis/. Accessed October 15, 2022. 2. Guier CP, Stokkermans TJ. Optic neuritis. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Updated September 25, 2022. www.ncbi.nlm.nih.gov/books/NBK557853/. Accessed October 15, 2022. 3. Balcer LJ. Clinical practice. Optic neuritis. N Engl J Med. 2006;354(12):1273-80. 4. Martínez-Lapiscina EH, Fraga-Pumar E, Pastor X, et al. Is the incidence of optic neuritis rising? evidence from an epidemiological study in Barcelona (Spain), 2008-2012. J Neurol. 2014;261(4):759-67. 5. Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65(6):727-32. 6. Beck RW, Cleary PA, Backlund JC, Optic Neuritis Study Group. The course of visual recovery after optic neuritis: experience of the Optic Neuritis Treatment Trial. Ophthalmology. 2020;127(4S):S174-81. 7. Keltner JL, Johnson CA, Cello KE, et al. Visual field profile of optic neuritis: a final follow-up report from the optic neuritis treatment trial from baseline through 15 years. Arch Ophthalmol. 2010;128(3):330-7. 8. Rosenthal JF, Hoffman BM, Tyor WR. CNS inflammatory demyelinating disorders: MS, NMOSD and MOG antibody associated disease. J Investig Med. 2020;68(2):321-30. 9. Weinshenker BG, Wingerchuk DM, Pittock SJ , et al. NMO-IgG: a specific biomarker for neuromyelitis optica. Dis Markers. 2006;22(4):197-206. 10. Kupersmith MJ, Alban T, Zeiffer B, et al. Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. 2002;125(4):812-22. 11. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald Criteria. Lancet Neurol 2018;17(2):162-73. 12. Sharma S, Leite MI. Typical and atypical optic neuritis – diagnosis and initial management. Eye News. Published June 1, 2016. www.eyenews.uk.com/education/top-tips/post/typical-and-atypical-optic-neuritis-diagnosis-and-initial-management. Accessed October 15, 2022. 13 Levin LA, Arnold AC. eds. Neuro-Ophthalmology: The Practical Guide. New York: Thieme; 2005. 14. Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of MS, third edition. Mult Scler. 2020;26(14):1816-21. 15. Hoftberger R, Lassmann H. Inflammatory demyelinating diseases of the central nervous system. Handb Clin Neurol. 2018;145:263-83. 16. Enders U, Lobb R, Pepinsky RB, et al. The role of the very late antigen-4 and its counterligand vascular cell adhesion molecule-1 in the pathogenesis of experimental autoimmune neuritis of the Lewis rat. Brain. 1998;121(Pt 7):1257-66. 17. Popescu BFG, Pirko I, Lucchinetti CF. Pathology of multiple sclerosis: where do we stand? Continuum (Minneap Minn). 2013;19(4):901-21. 18. Michel L, Touil H, Pikor JL, et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol. 2015;6:636. 19. Waksman BH. The distribution of experimental auto-allergic lesions: its relation to the distribution of small veins. Am J Pathol. 1960;37(6):673-93. 20. Tafti D, Ehsan M, Xixi KL. Multiple sclerosis. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing. Last updated September 7, 2022. www.ncbi.nlm.nih.gov/books/NBK557853/. Accessed October 15, 2022. 21. Rash JE, Yasumura T, Hudson CS, et al. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci USA. 1998;95(20):11981-6. 22. Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA. 2011;108(9):3701-6. 23. Jasiak-Zatonska M, Kalinowska-Lyszczarz A, Michalak S, et al. The immunology of neuromyelitis optica-current knowledge, clinical implications, controversies and future perspectives. Int J Mol Sci. 2016;17:273. 24. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-89. 25. Marrie RA, Gryba C. The incidence and prevalence of neuromyelitis optica. Int J MS Care. 2013;15(3):113-8. 26. Price CJ, Hoyda TD, Ferguson AV. The area postrema: a brain monitor and integrator of systemic autonomic state. Neuroscientist. 2008;14(2):182-94. 27. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicenter study of 175 patients. J Neuroinflammation. 2012;9:14. 28. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-14. 29. Xue T, Yu J, Chen S, et al. Different targets of monoclonal antibodies in neuromyelitis optica spectrum disorders: a meta-analysis evidenced from randomized controlled trials. Front Neurol. 2020;11:604445. 30. Kwon YN, Waters PJ, Kim M. Peripherally derived macrophages as major phagocytes in MOG encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. 2019;6(5):e600. 31. Pohl D, Alper G, Haren KV, et al. Acute disseminated encephalomyelitis: Updates on an inflammatory CNS syndrome. Neurology. 2016:87(9 Suppl 2):S38-45. |
