 |
A 35-year-old Caucasian female presented to our clinic seeking a second opinion for gradual, progressive, painless loss of vision in both eyes, which started about six years earlier. She provided exam notes from her initial exam six years earlier, and at that time, her vision was documented at 20/30 OD and 20/25 OS. She was taking no medications, had no known systemic medical conditions. Her past ocular history was otherwise unremarkable and there was no family history of ocular disease.
Upon examination, her best corrected visual acuity was 20/100 in each eye. Confrontation visual fields were full to finger counting in both eyes; pupils were equally round and reactive to light with no afferent pupillary defect. The slit lamp exam was unremarkable, and intraocular pressure was 16mm Hg OD and 14mm Hg OS.
On dilated fundus exam the optic nerve, retinal vasculature and peripheral retina were within normal limits in both eyes. In the central macula of each eye was a circular, well-circumscribed area of granular-appearing atrophy, with no drusen, edema or flecks (Figures 1a and 1b).
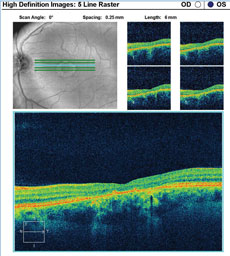
A fundus autofluorescence of the macula is available for review (Figures 2a and 2b). An SD-OCT image is also provided (Figures 3a and 3b).
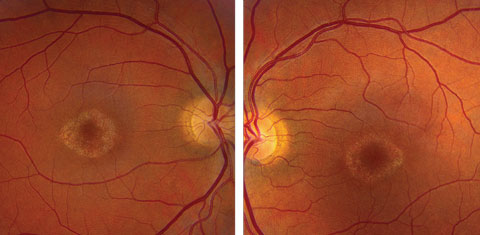 |
| Click image to enlarge. Figs. 1a and 1b. Fundus photos of the right and left eye of our patient. What do the changes in her macula represent? |
Take the Quiz
1. How do the fundus photos correlate to the OCT images?a. The OCT images show parafoveal atrophy of the outer retina and RPE in the areas of hypopigmented geographic atrophy.
b. The OCT images show subretinal deposits correlating to vitelliform lesions in the photos.
c. The OCT images show collapsed drusen in areas of choroidal atrophy in the photos.
d. The OCT shows blockage of the choroid.
2. What is the likely etiology?
a. Stargardt’s macular dystrophy.
b. Geographic atrophy from macular degeneration.
c. Central areolar choroidal dystrophy.
d. North Carolina macular dystrophy.
3. What is the typical inheritance pattern of this condition?
a. Autosomal dominant.
b. Autosomal recessive.
c. X-linked recessive.
d. Sporadic.
4. What additional testing would be most helpful to establish the correct diagnosis?
a. Fluorescein angiography.
b. Electrooculogram.
c. Genetic testing.
d. Macula risk test.
For answers, see below.
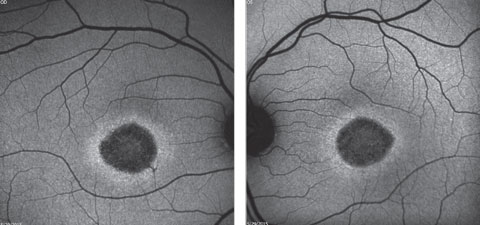 |
| Click image to enlarge. Figs. 2a and 2b. Fundus autofluorescence of the right and left eye. How do these correlate with the fundus images? |
Diagnosis
The maculas in both eyes have a characteristic “beaten metal” appearance. As a result, the initial doctors who saw her had a high suspicion for Stargardt’s disease. An electroretinogram (ERG) was done and showed normal rod response and mixed cone-rod response, while the multifocal ERG was depressed centrally, indicating localized retinal dysfunction. In an attempt to confirm the diagnosis of Stargardt’s disease, genetic testing was conducted, looking specifically for the ABCA4 gene mutation. Surprisingly, the test came back negative. The negative ABCA4 result coupled with the clinical absence of “flecks” made the diagnosis of Stargardt’s less likely, but still possible.ABCA4 gene mutation is the most common cause of autosomal recessive (AR) retinal disease, accounting for 95% of Stargardt’s disease, 30% to 50% of cone rod dystrophy and 8% of AR retinitis pigmentosa.1 There have been more than 250 different disease-causing alleles identified in ABCA4, though other locations have been implicated for Stargardt’s, including ELOVL4, PRPH2 and BEST1.1
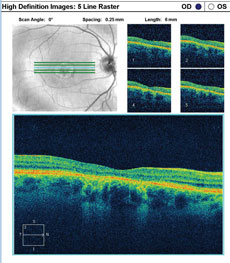 |
| Click images to enlarge. Figs. 3a and 3b. SD-OCT images show the patient’s retinal state in both the right and left eye. |
 |
With the negative ABCA4 gene mutation, we made the tentative diagnosis of central areolar choroidal dystrophy (CACD) based on the clinical appearance of the lesions, age of onset, normal full field electroretinogram and negative genetic testing for Stargardt’s disease.
The gradual decrease in vision with CACD usually begins in the third decade of life, with characteristic bilateral, solitary, well circumscribed central lesions that have nonspecific granularity within them.2 The central geographic atrophy that is seen in CACD is also distinguished by the absence of surrounding drusen and flecks which are seen in similar macular diseases.2 Often, patients will stabilize at 20/100 to 20/200 until the seventh or eighth decade of life.2
CACD was first described in 1884 and it is primarily inherited in an autosomal dominant fashion, though autosomal recessive cases have been occasionally reported.3 The most commonly associated mutation occurs on the PRPH2/RDS gene, and a wide range of disease severity and nonpenetrance can be observed, perhaps representing variants of AR or sporadic inheritance.4 These cases of low penetrance and milder disease can be found in 20% of CACD patients, often resulting in a misdiagnosis of geographic atrophy secondary to early onset AMD, which enlarges much more quickly than CACD related atrophy.4
In cases such as this, with symmetrical and bilateral geographic atrophy in the absence of drusen in a young patient, genetic testing may be beneficial to establish the correct diagnosis and possibly provide an opportunity for genetic counseling.
After discussing these findings with our patient, she expressed an interest in testing for as many possible inherited macular dystrophies as possible with the hopes of obtaining a conclusive diagnosis. She was also interested in possibly joining any future clinical trials on potential treatments.
The patient’s blood was drawn and sent for the comprehensive 127 gene Retinal Dystrophy Panel through the Casey Eye Institute Laboratory at Oregon Health and Science University. The test is listed at $2,500 and is almost always paid out of pocket, but for patients seeking a definitive cause for their visual impairment, this option is attractive to attain answers and closure.
A full list of available tests is available at ohsu.edu, along with instructions for specimen requisition and the relevant consent and payment forms. In most cases, the test results are available within one to three months.
The PRPH2/RDS and GUCY2D genes have been implicated in CACD and are included in the panel that will be run for this patient.4,5 She will be contacted with the results and notified of any clinical trials for which she may qualify. We plan to monitor her condition yearly for progression and updates on possible trials.
Dr. Hammond is an optometric resident at the Bascom Palmer Eye Institute in Miami.
|
1. Sohn E, Mullins R, Stone E. Macular Dystrophies. In: Ryan SJ ed. Retina. Vol 2, ed. V. St Louis:Mosby; 2013:864-74. 2. Yannuzzi LA. Cetntral Areolar Choroidal Dystrophy. The Retinal Atlas. 1st ed. Saunders Elsevier; 2010. 80. 3. Genead M, Fishman G, Grover S. Hereditary Choroidal Diseases. In: Ryan SJ ed. Retina. Vol 2, ed. V. St Louis: Mosby; 2013;891-2. 4. Boon C, Klevering B, Cremers F, et al. Central areolar choroidal dystrophy. Ophthalmology. 2009;116:771–82. 5. Hughes A, Meng W, Lotery A, Bradley D. A Novel GUCY2D Mutation, V933A, causes central areolar choroidal dystrophy. Invest Ophthalmol Vis Sci. 2012;53:4748–53. |
