 |
A 64-year-old male presented to the clinic for a second opinion after he failed the binocular vision test for his commercial driver’s license. The issue was preventing him from employment. He had an observable eye turn; when asked about it, he mentioned that he knew his eyelids had been “droopy” since childhood and that other members of his immediate family (father and brother) had the same thing. He denied variability of appearance or function.
The practitioner that had evaluated him initially (the first opinion), identified the issue and referred him to neuro-ophthalmology but he did not follow through. He had no other systemic disease, was taking no meds and did not indicate any allergies.
Diagnostic Data
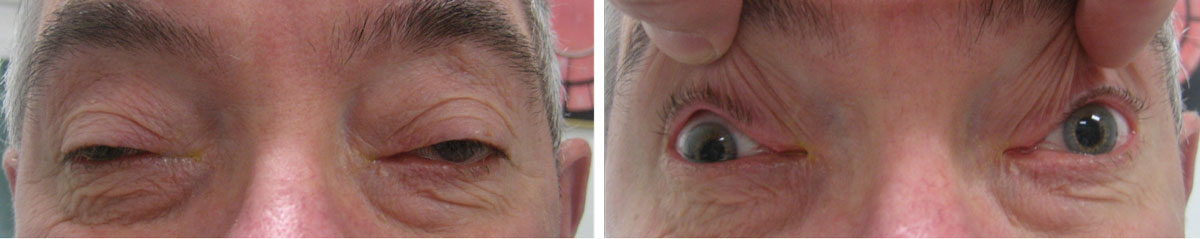
His best-corrected entering visual acuities through correction of –6.50 -2.00X180/+2.50 were 20/200 OD and 20/25 OS at distance and near with no improvement upon pinhole. The pertinent external findings are demonstrated in the photographs. Confrontation visual fields were full, less some lid-position superior constriction, and there was no afferent pupillary defect.
Cover test revealed a commitment 35 prism diopter right constant exotropia with steady temporal eccentric fixation, harmonious anomalous retinal correspondence, no stereo, a deep suppressive adaptation with severe amblyopia.
Extraocular muscle motilities demonstrated limited upgaze OU with a mild adduction deficit OD. Refraction demonstrated negligible changes with minimal improvement in acuities. Biomicroscopy uncovered normal anterior segment structures and open angles OU. Goldmann intraocular pressures measured 11mm Hg OU.
The dilated fundus examination revealed mild myopic conus OU (staphyloma posticum) with no evidence of choroidal neovascularization and normal peripheries.
 |
|
What do these findings suggest about the patient? How would you approach management? Click image to enlarge. |
Additional Testing
The search for other relevant factors included asking about difficulty swallowing (dysphagia) and checking for limb weakness or fatigability. Eyelid closure strength was compared between the two eyes to determine if the process was binocular or asymmetric. An ice pack test or sleep test could be done in the office to test for myasthenia gravis. This test uses cold to decrease the effectiveness of acetylcholinesterase (the biochemical agent that breaks down the neurotransmitter acetylcholine), allowing it to stay in the synaptic cleft longer, reducing lid symptoms and signs in myasthenic patients. The sleep test permits acetylcholine to build up in the cleft while the eyelids are closed. Function improves upon waking, as the neuromuscular junction is full of the neurotransmitter.
The pupils could be measured to rule out anisocoria and potential Horner’s syndrome. Laser interferometry could be completed to uncover the best potential acuity of the right eye and confirm loss of acuity via strabismic amblyopia.
Discussion
The diagnosis in this month’s case is bilateral upper eyelid ptosis, poor levator function and globally reduced eye movements due to mitochondrial myopathy or chronic progressive external ophthalmoplegia (CPEO).
CPEO is defined as an external ophthalmoplegia secondary to mitochondrial myopathy that is slowly progressive.1-7 Typically, there is symmetric blepharoptosis and limitation of ocular motility in all directions of gaze.1-4 Cases without ptosis are uncommon but have been reported.2
In general, mitochondrial diseases are a large group of disorders resulting from mutations of nuclear DNA and mitochondrial DNA (mtDNA).1-4,7 Mitochondrial disorders represent a clinically, biochemically and genetically heterogeneous group of diseases associated with dysfunction of the oxidative phosphorylation system and pyruvate dehydrogenase complex.7 The condition may occur in isolation as the individual matures or may occur in younger patients as one feature of a systemic multi-organ disorder.2-7
Prevalence is estimated at one in 5,000.7 Such conditions may manifest at any age through late-adulthood.7 When clinical signs and symptoms begin, they may present as acute manifestations or as a series of chronic progressive issues.7 Virtually any organ may be impaired, but the organs with the highest energetic demands are the ones that are most frequently involved (brain, muscle, heart and liver).7
The CPEO variant demonstrates ptosis from levator palpebrae superioris weakness with or preceding ophthalmoplegia. This represents the earliest clinical feature of the disease.1-4 CPEO patients often present with multiple manifestations, including myopathies and multiple system disorders; this is known as CPEO-plus.2
Non-ocular skeletal myopathy, fatigue, neurologic sequelae (peripheral neuropathy, migraine headaches, seizures, stroke-like episodes, ataxia, sensorineural hearing loss), skin findings, gastrointestinal involvement (dysphagia, liver failure, diarrhea, constipation), neuropsychiatric abnormalities, endocrinopathies and cardiac disorders (cardiomyopathy and conduction defects) have all been reported as potential sequelae.1-4
The combination of CPEO with systemic symptoms is often given syndromic names, such as Kearns-Sayre syndrome (onset in childhood with progressive external ophthalmoplegia, cardiac conduction defects, pigmentary retinopathy, short stature, sensorineural hearing loss, ataxia, dementia and migraines), myoclonus epilepsy (ragged red fibers with onset in late childhood to adolescence; myoclonic and generalized seizures, ataxia, generalized muscle weakness and progressive external ophthalmoplegia), Leigh syndrome (onset in infancy; ataxia, seizures, sensorineural hearing loss, dysphagia, dysarthria, progressive external ophthalmoplegia), Pearson syndrome (onset in infancy; anemia, pancreatic failure, progressive external ophthalmoplegia, cardiac conduction defects, and pigmentary retinopathy) and Alpers syndrome (onset in infancy; seizures, liver dysfunction, dementia, spasticity, central blindness).2
Establishing a specific diagnosis often requires extensive clinical and laboratory evaluation because so many things must be ruled out.1-4 A thorough family history may reveal the presence of consistent findings amongst those most susceptible to the genetic pattern of penetration of a mitochondrial disease.5 Mitochondrial diseases are inherited as Mendelian disorders characterized by disturbed mtDNA maintenance.5 mtDNA is a small, circular extranuclear chromosome encoding essential components of the respiratory chain.6 mtDNA-related syndromes are divided into two groups: mitochondrial encephalomyopathies, characterized by the presence of ragged-red fibers, or “pure” encephalopathies with no gross morphological abnormalities in muscle.6
The first group includes myoclonic epilepsy with ragged-red fibers, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes, Kearns-Sayre syndrome, chronic progressive external ophthalmoplegia and maternally inherited myopathy and cardiomyopathy.6 The second group includes Leber’s hereditary optic neuroretinopathy and ataxia-retinitis pigmentosa-dementia complex.6
Signaling errors lead to mtDNA depletion and or accumulations of mtDNA deletions in critical tissues.5,6 The genes involved encode for proteins that disrupt normal mtDNA replication, maintenance, nucleotide supply, mitochondrial dynamics and quality control.5 In many cases, mutations in these faulty genes lead to profound phenotypes.5
The differential diagnosis includes blepharochalasis, blepharophimosis (blepharophimosis, ptosis and epicanthus inversus syndrome, or BPES), congenital fibrosis of the extraocular muscles (a group of conditions characterized by congenital paralytic strabismus secondary to restrictive ophthalmoplegia, often with accompanying ptosis), Horner’s syndrome (sympathetic interruption; ptosis, meiosis and anhidrosis), Marcus-Gunn jaw wink (ptosis increased by moving the jaw towards the affected side; a synkinesis resulting from aberrant connection between the motor branches CNV which innervates the pterygoid muscles and fibers of the oculomotor nerve), cranial nerve III palsy and Duane’s retraction syndrome (aberrant regeneration producing limited gaze palsies and co-contracture of medial and lateral rectus creating enophthalmos and bilateral ptosis).8-10
Case Management
Workup for suspected cases is best completed by neuro-ophthalmology. Testing includes a detailed history, comprehensive clinical examination, specialized examinations (cardiology, visual fundus examination, brain imaging, EMG), laboratory testing of body fluids (lactate, amino acids, organic acids) and analysis of samples of muscle, skin, and liver.7 Normal lactate levels in blood does not exclude the possibility of mitochondrial disease.7
Treatment involves counseling and supportive measures to protect the ocular surface and improve cosmesis and functioning with lid lifting procedures such a frontalis suspension or levator resection.8 Treatment is not possible for this genetic condition.1-7 Due to possible weak orbicularis function, ptosis surgery must be approached with caution as poor postoperative lid position may result in corneal exposure.8
Genetic testing and counseling is recommended for those patients who may be planning on having children.1-7 BPES is usually inherited in an autosomal dominant manner; autosomal recessive inheritance has been reported in one consanguineous family.9 In cases of autosomal dominant inheritance, each child of an individual with BPES has a 50% chance of inheriting the FOXL2 pathogenic variant.9 Prenatal testing for pregnancies at increased risk is possible.9
The prognosis for these cases is reasonable so long as proper timely surgical solutions can be obtained. In cases where amblyopia of disuse occurs secondary to the vision being blocked, the cosmetic prognosis may be improved but the functional result is usually poor.
Dr. Gurwood thanks Marc Myers, OD, for contributing this case.
Dr. Gurwood is a professor of clinical sciences at The Eye Institute of the Pennsylvania College of Optometry at Salus University. He is a co-chief of Primary Care Suite 3. He is attending medical staff in the department of ophthalmology at Albert Einstein Medical Center, Philadelphia. He has no financial interests to disclose.
1. Murdock J, Thyparampil PJ, Yen MT. Late-Onset Development of Eyelid Ptosis in Chronic Progressive External Ophthalmoplegia: A 30-Year Follow-up. Neuroophthalmology. 2016;40(1):44-46. |
