 |
A 21-year-old Caucasian myopic female presented with a two-week history of acute onset painless central vision loss in her right eye. She denied flashing lights, floaters, redness, pain and photophobia. Her past medical, ocular and family histories were all unremarkable. She was a college student studying pre-law and endorsed significant stress levels in preparing for final exams. A thorough review of systems only elicited outbreak of cold sores within two weeks of symptom onset. Posterior segment imaging can be found below.
Visual acuity (VA) was 20/100 with pinhole improvement to 20/70 OD and 20/20 OS. IOP was 15mm Hg OD and 13mm Hg OS. Confrontation visual fields were full to finger counting and there was no relative afferent pupillary defect. Anterior segment examination was normal and notably negative for anterior chamber or vitreous cell.
Take the Retina Quiz
1. Which of the following regarding interpretation of the multimodal imaging is false?
a. There are multifocal gray-white lesions in the right macula by fundus photos.
b. There is multifocal hyperautofluorescence in the right macula by fundus autofluorescence.
c. There is multifocal hyperfluorescence in the right macula by fluorescein angiography.
d. There is subretinal fluid in the right macula on OCT.
2. What is the most likely diagnosis?
a. Multifocal choroiditis with panuveitis.
b. Multiple evanescent white dot syndrome.
c. Punctate inner choroidopathy.
d. Presumed ocular histoplasmosis syndrome.
3. Which of the following is NOT typical of the demographic for this disease entity?
a. Asian descent.
b. Female gender.
c. Myopic refractive error.
d. All of the above are typical.
4. What is the average visual prognosis for this condition?
a. 20/20 or better.
b. 20/40 or better.
c. 20/200 or worse.
d. Hand motions or worse.
5. Which of the following is a reasonable treatment option for this patient?
a. Intravitreal anti-vascular endothelial growth factor (VEGF) agent.
b. Observation.
c. Systemic corticosteroid.
d. All of the above.
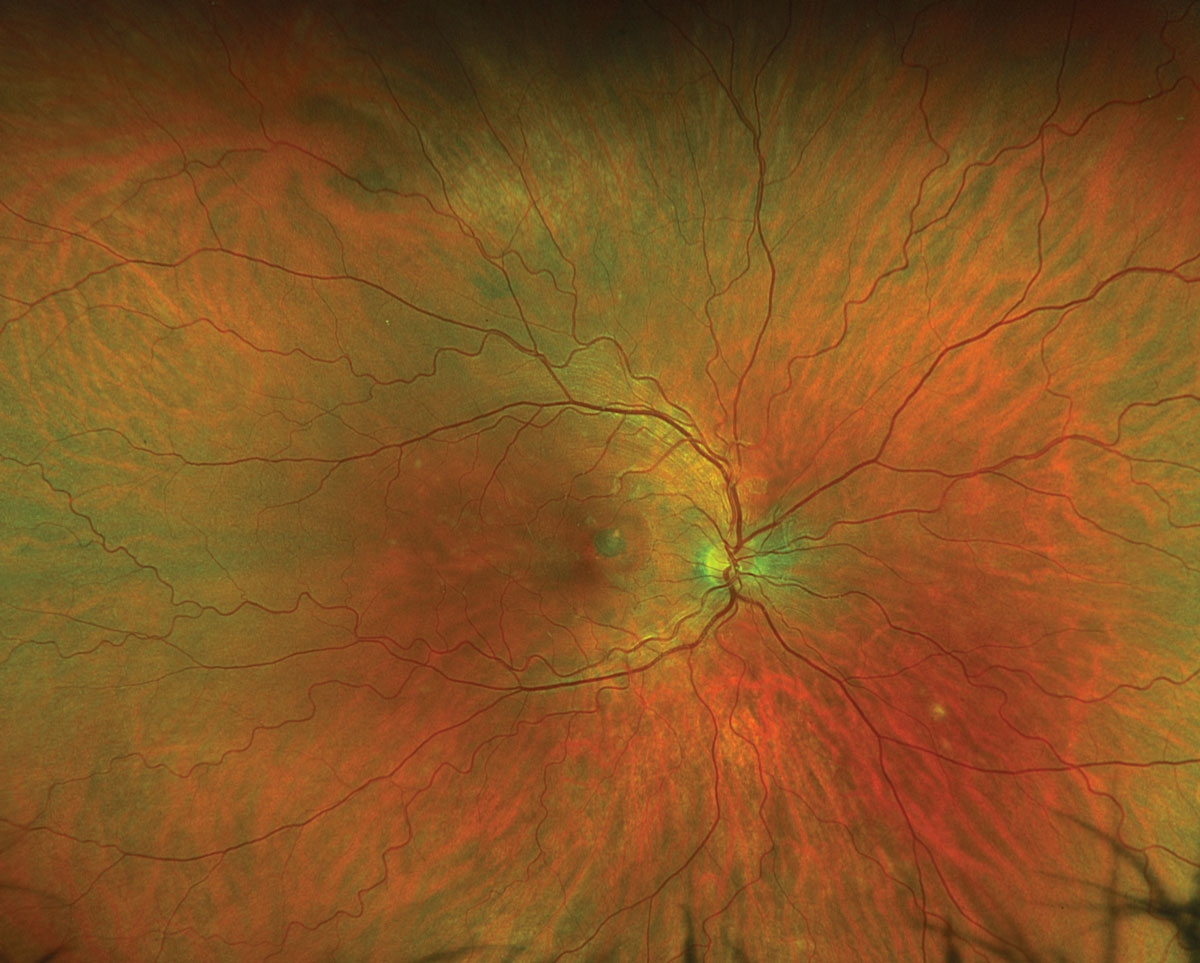 |
Fig. 1. Optos ultra-widefield fundus photograph of the right eye. Click image to enlarge. |
Diagnosis
Fundus exam revealed a parafoveal hyperpigmented macular lesion with faint punctate white spots throughout the macula (Figure 1). The parafoveal hyperpigmented lesion demonstrated gross hypo-autofluorescence (AF) with a faint ring of hyper-AF, while the macular white spots all demonstrated hyper-AF on fundus AF (Figure 2). OCT demonstrated multifocal photoreceptor loss involving the fovea and a parafoveal hyperreflective subretinal lesion without evidence of exudation (Figure 3). Fluorescein angiography showed staining of the parafoveal lesion as well as the white macular dots seen clinically; there was notably no leakage seen (Figure 4). Indocyanine green angiography demonstrated hypocyanescence of some of the macular lesions in the early and late frames. Serologies for syphilis, tuberculosis, sarcoidosis and general blood chemistries were negative. A clinical diagnosis of punctate inner choroidopathy (PIC) was made.
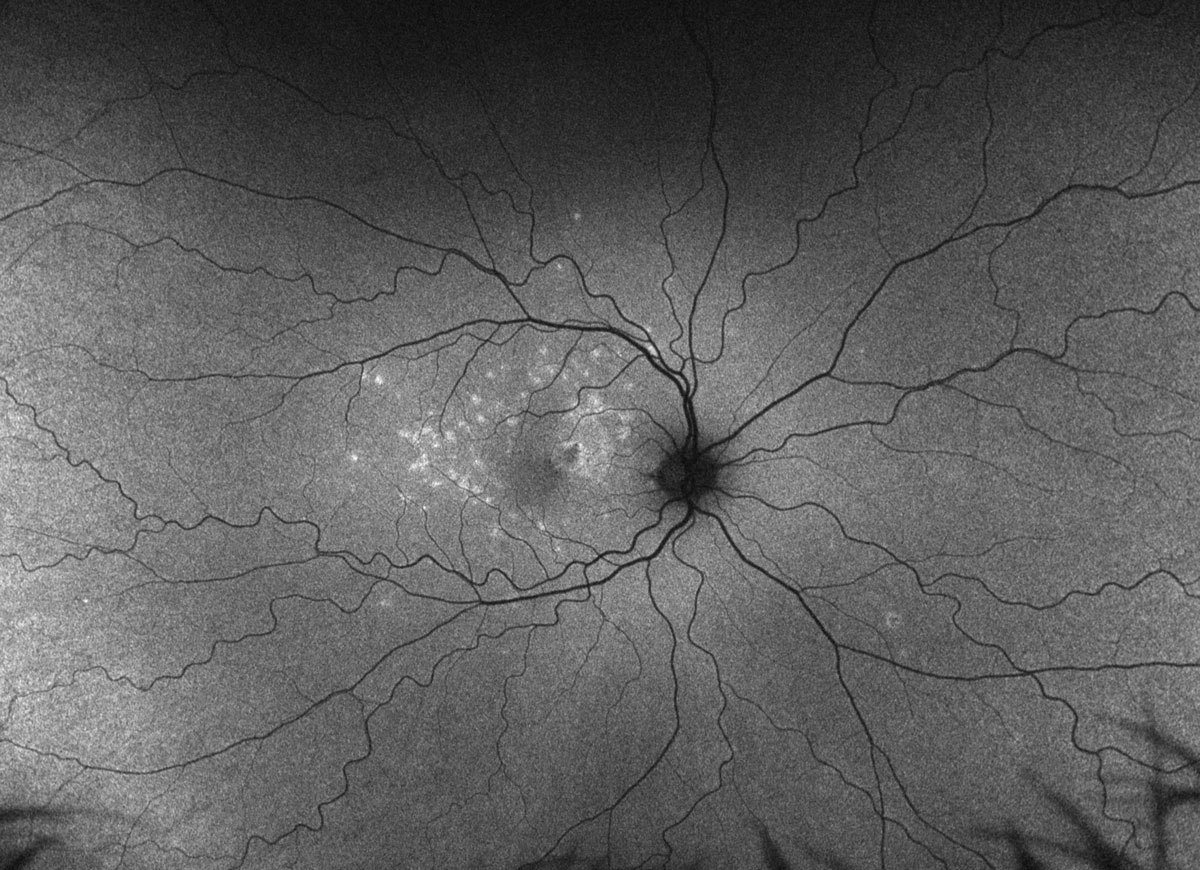 |
Fig. 2. Optos ultra-widefield fundus autofluorescence of the right eye. Click image to enlarge. |
Discussion
First described in 1984, PIC is a rare inflammatory condition typically affecting young, white (97%), myopic (85%) women (90%).1,2 Presentation is often within the second through fifth decades of life with a mean age at diagnosis in the 30s (median 30 years old) and mean refractive error of -4.5D (median -7D), with only 1% of patients being hyperopic.1,3 Typical symptoms includes scotomas, photopsias, metamorphopsias and blurred vision.1,4 Pathophysiology is thought to be autoimmune, and the presence or family history of autoimmune disease may be a predisposing risk factor; furthermore, a direct familial association of PIC has been demonstrated.5
It is believed that PIC, multifocal choroiditis (MFC) and multifocal choroiditis with panuveitis (MCP) all lie on the same spectrum of disease, or at least share a similar pathogenesis.3,4 While distinction between PIC and MFC can be challenging, clinical presentation of PIC generally includes multiple round, gray/yellow/white lesions (100µm to 300µm in diameter) of the inner choroid and retinal pigment epithelium (RPE) that are distributed throughout the posterior pole, and often evolves to bilateral involvement.1,2,4 Lesions in MFC tend to be larger in size and extramacular in location, and MCP shows a similar fundus appearance to MFC in the setting of panuveitis, which is notably absent in PIC.3,4 Once quiescent, acute inflammatory lesions either produce foci of chorioretinal atrophy with or without pigmentation or resolve entirely without atrophic changes.3 The resultant fundus appearance may be indistinguishable between PIC, MFC and MCP, and also presumed ocular histoplasmosis syndrome. Of note, the absence of vitritis does not necessarily exclude MCP; conversely, the presence of vitritis does not exclude presumed ocular histoplasmosis.4 Sequelae of chronic inflammation include macular neovascular membranes and subretinal fibrosis which can impact final VA.3
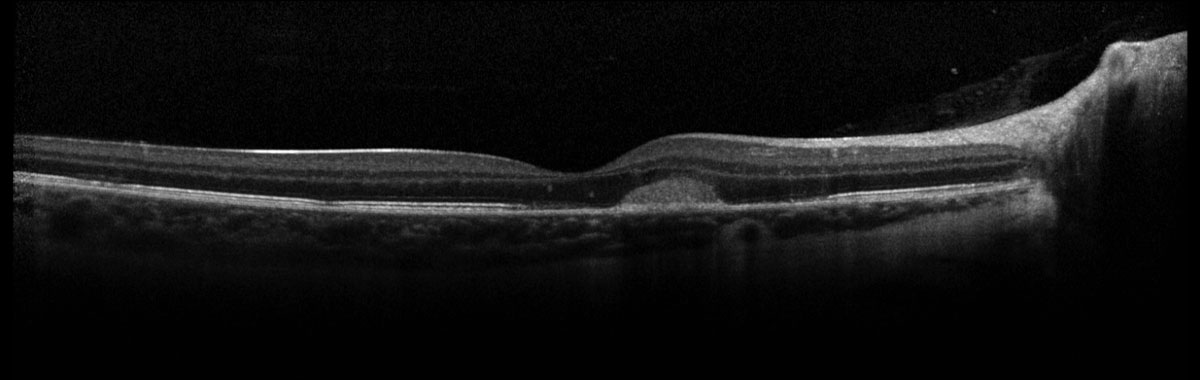 |
|
Fig. 3. Spectralis Heidelberg OCT of the right eye. Click image to enlarge. |
Multimodal Imaging
This is important when evaluating uveitic entities to best characterize the structures involved and extent of disease dissemination. Fundus AF will show hyper-AF of acute lesions and hypo-AF corresponding with subsequent atrophic changes.5 Spectral domain OCT is limited in its ability to image the choroid, but may show acute hyperreflective opacities in the sub-RPE and choroidal space, multifocal outer-retinal and RPE atrophy, choroidal thickening due to cellular infiltration, subretinal fibrosis, macular neovascular membranes formation and subretinal fluid.3,5 Fluorescein angiography classically shows early hyperfluorescence with late staining and indocyanine green angiography shows early and late hypocyanescence with sporadic hypercyanescence of blood vessel walls, suggestive of a vasculitic component in some patients.1-3,5
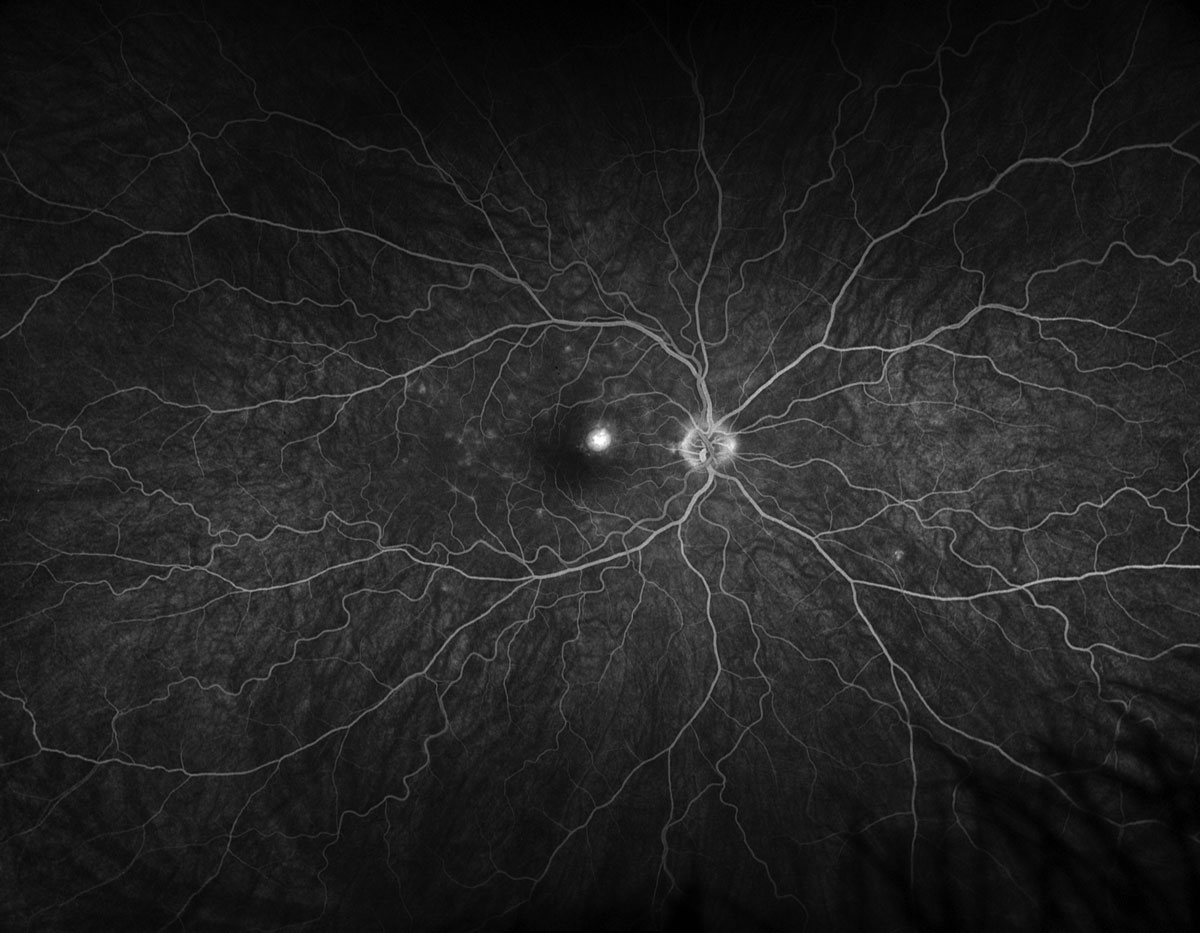 |
Fig. 4. Optos ultra-widefield fluorescein angiography late phase of the right eye. Click image to enlarge. |
Treatment
Systemic, local and periocular corticosteroids have been used for acute inflammatory lesions and regression of acute macular neovascular membranes, and immunomodulatory therapy has been used in small sample sizes to successfully mitigate disease recurrence.4 Macular neovascular membrane management has evolved from photodynamic therapy to intravitreal anti-VEGF drugs with successful regression of macular neovascular membranes.3,4 All therapies carry their own risk/reward profile that should be considered and reviewed in the collaborative decision-making between physician and patient.
Prognosis
Final visual acuity and prognosis in PIC are generally favorable in the absence of macular neovascular membranes, though 40% to 83% of patients either present with or develop macular neovascular membrane lesions at some point in their disease course.1,3-6 VA outcomes range from 20/40 or better in as many as 66% to 77% of patients, to 20/200 or worse in 15% to 20% (largely due to fovea-involving macular neovascular membrane and subretinal fibrosis).1,3,6
We felt that our patient presented more subacutely given the presence of a moderately pigmented subretinal macular neovascular membrane adjacent to the fovea. All treatment options were discussed and the collective decision was made to observe closely. The macular dots slowly regressed clinically and on fundus AF in the absence of treatment and the macular neovascular membranes remained stable on OCT with no evidence of exudation at follow-up four months later. She will continue to be monitored for recurrence or evolution to bilaterality.
Retina Quiz Answers
1: d, 2: c, 3: a, 4: b, 5: d
Dr. Aboumourad currently practices at Bascom Palmer Eye Institute in Miami. Neither has any financial disclosures.
Dr. Dunbar is the director of optometric services and optometry residency supervisor at the Bascom Palmer Eye Institute at the University of Miami. He is a founding member of the Optometric Glaucoma Society and the Optometric Retina Society. Dr. Dunbar is a consultant for Carl Zeiss Meditec, Allergan, Regeneron and Genentech.
1. Amer R, Lois N. Punctate inner choroidopathy. Surv Ophthalmol. 2011;56(1):36-53. 2. Watzke RC, Packer AJ, Folk JC, et al. Punctate inner choroidopathy. Am J Ophthalmol. 1984;98(5):572-84. 3. Campos J, Campos A, Mendes S, et al. Punctate inner choroidopathy: a systematic review. Med Hypothesis Discov Innov Ophthalmol. 2014;3(3):76-82. 4. Agarwal A, Gass JDM. Gass’ Atlas of Macular Diseases, 5th ed. London: London: Elsevier - Health Sciences Division. 2011. 5. Cicinelli MV, Ramtohul P, Marchese A, et al. Latest advances in white spot syndromes: new findings and interpretations. Prog Retin Eye Res. 2023;97:101207. 6. Essex RW, Wong J, Fraser-Bell S, et al. Punctate inner choroidopathy: clinical features and outcomes. Arch Ophthalmol. 2010;128(8):982-7. |
