
A key element in triaging retinal disease is making a definitive diagnosis. Many, if not most, retinal diseases are either self-limiting or non-treatableboth of which can be managed in an advanced primary eye care office.
Here, a case of multiple evanescent white dot syndrome (MEWDS) is presented. An astute clinician is capable of making the initial suspected diagnosis. Scope of practice certifications are broadening, and some states are including flourescein angiography (FA), which in this instance, was integral to the diagnosis.
This case is an example of a condition that can be diagnosed and managed within an advanced primary care optometric office, and certainly within a mixed M.D./O.D. practice.
Once the initial diagnosis is confirmed through specialized testing, either at an advanced optometric eye care office or by referral to a retinal specialist, such self-limiting conditions can easily be monitored within an optometric setting.
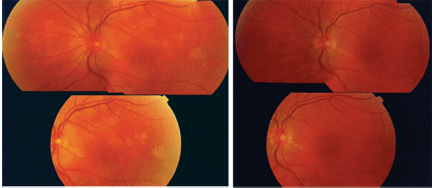
A fundus photo of the patients left eye demonstrates the active phase of MEWDS (left). An image taken two weeks later shows the conditions resolution (right).
History
Beth, a 55-year-old white female, presented for a routine eye exam to discuss the feasibility of contact lenses for presbyopia. She worked as a cosmetologist and was in good systemic health. The only medication that she reported was estrogen replacement. Her refraction revealed emmetropia O.U. with presbyopia during an otherwise-normal ophthalmic exam. At the conclusion of the exam and contact lens fitting, she was discharged as a happy monovision patient with one reading contact lens on her left eye.
Four months later, Beth called the office with concerns about her monovision contact lens. Beth said that she could no longer focus on anything with her reading eye, even after cleaning the lens or inserting a new one, and believed that she needed a stronger lens. The contact lens technician told her that it would be unusual for such a drastic change to occur over such a short time period with a non-painful, non-symptomatic eye. Beth was scheduled for an appointment later that same day.
Diagnostic Data

A Humphrey visual field 30-2 revealed a temporal field defect O.S.
Upon examination, Beths uncorrected distance acuity was 20/20 O.D., and her corrected near acuity was J3.
A detailed history revealed decreased vision O.S. for approximately three days. It seemed to be of gradual onset, and Beth also reported a temporal visual field defect O.S. that was confirmed by confrontational fields.
She reported that she had suffered from mild flu-like symptoms within the past two weeks, but had dismissed it as a mild 24-hour virus.
At this point, the patient was asked to remove the reading contact to determine her best-corrected distance acuity O.S. A minimal refractive error revealed best-corrected visual acuity of 20/30 O.S. Pupils and extraocular motilities were normal. No afferent pupillary defect was noted. Amsler grid findings were normal O.D., but her left eye demonstrated multiple areas of metamorphopsia.
The slit lamp examination was within normal limits O.U. Goldmann tonometry revealed an IOP of 13mm Hg O.U. Dilated fundus examination was normal O.D. and symmetrical discs with 0.2/0.2 cups were noted O.U. Critical examination of the left optic nerve revealed no signs of disc edema.
The most discerning finding of the fundus exam was the granular appearance of the left macula and the scattered white spots in paramacular areas. This finding was difficult to visualize ophthalmoscopically, and no vessel occlusions were noted. A Humphrey visual field 30-2 was ordered and revealed an enlarged blind spot that produced a nearly complete temporal field defect O.S. OCT testing was relatively unremarkable and showed only minimal macular thickening O.S.
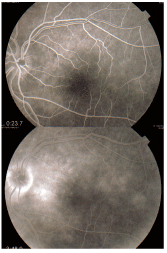
Beths FA displayed the classic wreath-like pattern of pinpoint white dots around the macula O.S.
Diagnosis
There was significant clinical evidence to support the diagnosis of a white dot syndromespecifically, MEWDS.
After consulting with an in-office M.D. colleague, we opted for an FA to complete the evaluation. The FA displayed the classic signs of multiple evanescent white dots and subtle RPE defects, which confirmed the diagnosis.
Treatment and Follow-Up
Due to the self-limiting nature of MEWDS, the patient was asked to return to the office six weeks later for monitoring. At that time, her best-corrected visual acuity was 20/20 O.D. and 20/20-2 O.S. Her ophthalmic examination was completely normal, including the funduscopic exam of the left eye.
Beth was pleased with her visual outcome and had a normal retinal exam, so she requested no repeat Humphrey visual field or FA testing due to financial concerns. At this point, she was discharged and asked to return in four to six months for monitoring, unless she experienced a change before that time.
Discussion
MEWDS was first described in 1984, and is classified as a variant of posterior uveitis along with the other white dot syndromes, such as acute posterior multifocal placoid pigment epitheliopathy, diffuse unilateral subacute neuroretinitis, multifocal choroiditis with panuveitis, birdshot chorioretinopathy and serpiginous choroiditis.1 MEWDS usually presents unilaterally and primarily affects people between the ages of 20 and 45. The syndrome has a greater predilection for females than males.2 And, up to 35% of cases report a systemic viral prodromein Beths case, the mild flu-like symptoms.
Otherwise, there appears to be no racial predilection or systemic links, though some reports suggest potential associations with the genetic marker HLA-B51 and the hepatitis B vaccine.2,3
Typically, the patient presents complaining of a unilateral decrease in vision, including temporal and paracentral scotomas. He or she may also report photopsia and dyschromatopsia. Visual acuity may vary between 20/20 and 20/400, depending on the severity of the syndrome. And, it is possible to have a relative afferent pupillary defect in cases with significant optic nerve involvement.
The anterior segment exam is generally normal with no inflammation in the anterior chamber. There may be rare to mild vitreous cells noted. Funduscopic examination may reveal a hyperemic and edematous optic disc. The characteristic appearance of this syndrome is the non-descript multiple small white dots scattered through the peripheral macula and around the optic nerve. These lesions are located at the outer retina or retina pigmented epithelium. The fovea may have an unusual orange-yellow color with granularity.4
Visual field testing will reveal an enlarged blind spot and other temporal and paracentral scotomas. FA will display early hyperfluorescence and late staining in a wreath-like pattern formed by the appearance of multiple white dots.1 In some cases, the disc may display hyperfluorescence in late phase. Indocyanine green angiography can reveal multiple hypofluorescent spots in the fundus and will confirm any choroidal involvement.5 Electroretinogram testing may show a reduced A-wave, indicating decreased photoreceptor function. Visually evoked cortical potential can create a delay in some cases, implicating optic nerve involvement.3
The acute phase of MEWDS begins to subside within one or two weeks. As the symptoms spontaneously resolve, so do the clinical signs. The only management protocol is to monitor for reoccurrence and ensure that there is no other etiology.
Visual prognosis is excellent; most patients recover normal vision within several weeks to months.3,4 A few patients have reported persistent symptoms of photopsia and dyschromatopsia, and rare cases of a permanent enlargement of the blind spot have been reported. Reoccurrence has also been reported, but this is rare.4
In the 22 years since MEWDS was first reported in the literature, the exact cause of this puzzling syndrome is still unknown. It is hypothesized to be an immune-mediated vascular inflammation that affects various retinal layers, including the choroid, RPE and photoreceptors.2,5 More recent clinical and angiographic studies posit that there may be multiple forms of MEWDS that affect various retinal layers and the choroid differently. Current research focuses on determining host predilection, immune-mediated mechanisms and genetic predispositions.5
All these varying factors most likely contribute to the interesting mix to this syndrome.
This is just one example of many self-limiting retinal conditions that we need to consider. As our profession continues to evolve with expanding scopes of practice, we need to broaden our knowledge of these conditions. Such uncommon, self-limiting, typically unilateral retinopathies can be diagnosed and managed in an advanced primary care setting, which saves the patient needless, stressful referrals to an unfamiliar specialist.
Dr. Foley is currently a partner at Eye Central with offices in
1. Jampol LM, Sieving PA, Pugh D, et al. Multiple evanescent white dot syndrome. I: Clinical findings. Arch Ophthalmol 1984 May;102(5):671-4.
2. Arbet TP. Multiple evanescent white dot syndrome. J Am Optom Assoc 1997 Dec;68(12):769-74.
3. Alexander LJ. Primary Care of the Posterior Segment. 3rd ed.
4. Quillen DA, Davis JB, Gottlieb JL, et al. The white dot syndromes. Am J Ophthalmol 2004 Mar;137(3):538-50.
5. Gross NE, Yannuzzi LA, Freund KB, et al. Multiple evanescent white dot syndrome. Arch Ophthalmol 2006 Apr;124(4):493-500.
