Making the DiagnosisExplore these other articles featured in our 9th Annual Diagnostic Skills & Techniques Issue.
|
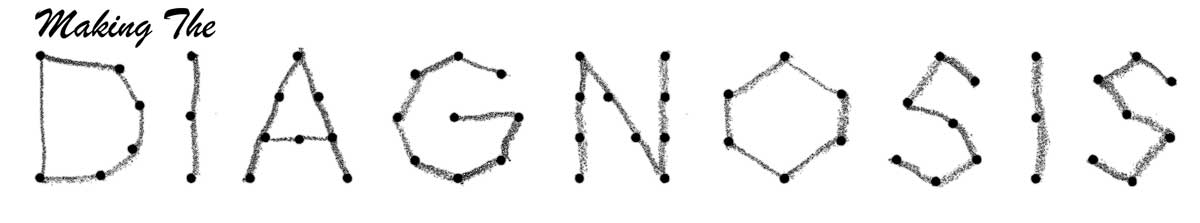
When a patient presents with what appears to be a medical emergency, you need to act fast to prevent the situation from escalating even further and potentially resulting in irreversible, devastating consequences. Some ophthalmic diseases are considered vision- and/or life-threatening, warranting their inclusion in the spectrum of medical emergencies or urgencies.
We three doctors—an optometrist, an ophthalmologist and an emergency medicine physician—are involved in different stages of the care such patients require. Here, we’ve pooled our collective expertise to outline a few of these conditions from the perspective of the entire management team to give you a quick reference guide to the workup, diagnostic clues and management course likely to occur for each.
 |
|
This patient with an intracranial mass seen on MRI presented with papilledema. The resolution is noted on OCT post-mass resection. Click image to enlarge. |
Papilledema
Defined as bilateral swelling of the optic nerve head (ONH) caused by increased intracranial pressure (ICP), papilledema can cause transient visual obscurations, visual field loss, pulse synchronous tinnitus, retrobulbar pain, diplopia, headache, nausea and vomiting. Optic nerve damage is caused by intraneuronal ischemia and axoplasmic stasis.1
History. Ask patients about vision changes, binocular diplopia, tinnitus, headache, focal paresthesia, focal weakness, ataxia, aphasia, fevers, chills, recent pregnancy, recent weight gain and history of cancer. Review their meds; oral contraceptives, tetracyclines, vitamin A derivatives and exogenous steroids (to name a few) have been known to cause secondary intracranial hypertension.
Examination. Papilledema is more commonly bilateral and symmetric but can occasionally present with asymmetric involvement. Perform a complete eye exam with attention to pupils to monitor for an afferent pupillary defect (APD), color plates and confrontation visual fields. Note that if both optic nerves are equally affected, an APD may be absent.
Evaluate extraocular motility to assess for a sixth nerve palsy, frequently seen in idiopathic intracranial hypertension (IIH). Perform a dilated fundus exam to assess the appearance of the optic nerves. There are varying degrees of disc elevation and blurred disc margins with or without associated peripapillary hemorrhages and cotton wool spots. Conduct visual field testing and optical coherence tomography (OCT) of the optic nerves. Consider fundus autofluorescence or B-scan imaging to rule out causes of pseudo-papilledema such as ONH drusen. Check blood pressure in all patients with bilateral optic nerve edema to rule out malignant hypertension.
Differential diagnosis. Causes of increased ICP include intracranial mass lesions, cerebral hemorrhage, cerebral edema, arteriovenous malformations, head trauma, cerebral venous sinus thrombosis, hydrocephalus, meningitis, encephalitis and IIH.
A study evaluating papilledema in the outpatient setting found that 87% of cases were caused by IIH and that 13% of patients had potentially life-threatening conditions, such as intracranial tumor, cerebral venous sinus thrombosis and granulomatous meningitis, 22% of whom had localizing neurologic signs.2 This demonstrates the importance of prompt neuroimaging to rule out a life-threatening complication.
Other causes of optic nerve swelling not associated with elevated ICP include hypertensive retinopathy, diabetic papillopathy and toxic, infectious and inflammatory optic neuropathy.
Workup. All patients with ONH edema require prompt neuroimaging, including magnetic resonance imaging (MRI) with and without contrast and magnetic resonance venogram. After neuroimaging has ruled out intracranial mass lesions, lumbar puncture should be performed to evaluate ICP and cerebrospinal fluid (CSF). Elevated ICP is defined as greater than 25cm of water. Elevation of CSF white blood cells or CSF protein may be suggestive of an infectious, inflammatory or neoplastic cause. IIH is a diagnosis of exclusion when ICP is elevated and the CSF profile is negative.
It can be challenging to obtain timely neuroimaging and neurology consultation in the outpatient setting; thus, to rule out life-threatening complications, referral to the emergency department (ED) is indicated. If possible, refer to a regional medical center with access to neurology, neuroradiology and neurosurgery.
Treatment. This depends on the underlying cause of papilledema. IIH is treated with weight loss and diuretics such as acetazolamide. In cases refractory to medical therapy, surgical intervention is warranted with either optic nerve sheath fenestration performed by an oculoplastic surgeon or neuro-ophthalmologist or a ventriculoperitoneal or lumboperitoneal shunt performed by a neurosurgeon. Choice of surgical intervention depends on whether the prominent symptoms are vision loss/changes or headache, respectively.
Intracranial mass lesions such as tumors, abscesses and hemorrhages warrant emergent neurosurgical consultation. Infectious conditions such as meningitis and encephalitis warrant evaluation by neurology and infectious disease specialists.
 |
|
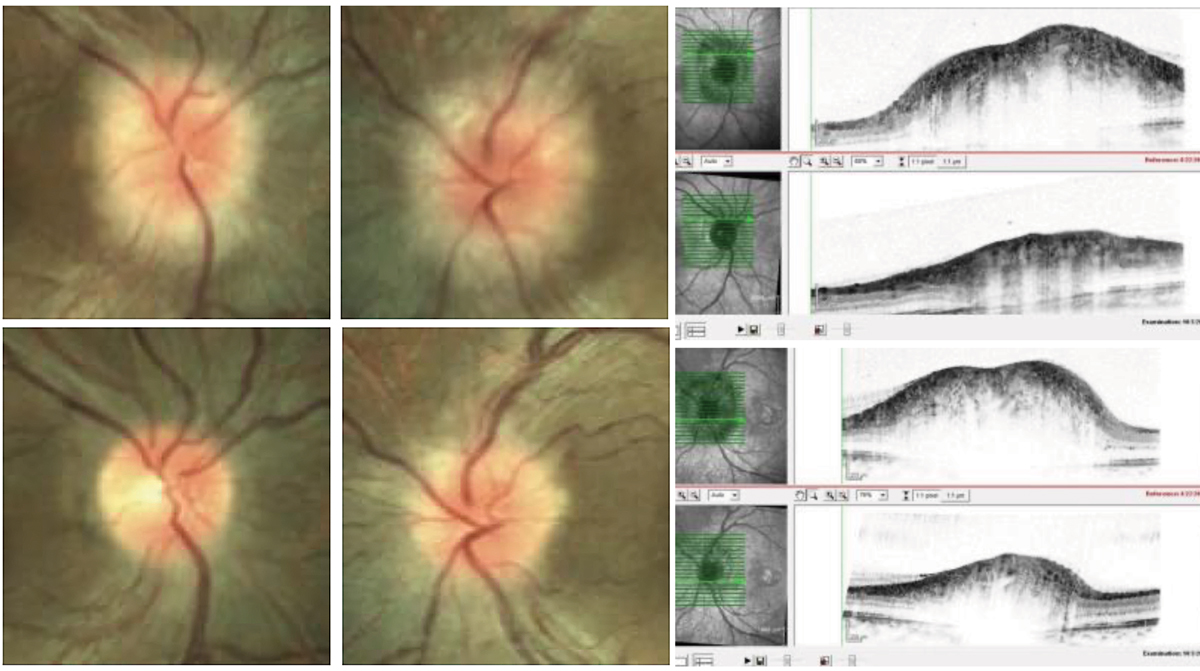
This patient presented with papilledema and was diagnosed with IIH following lumbar puncture and negative MRI/MRA. After treatment with oral acetazolamide, complete resolution of disc edema is noted with some degree of secondary disc pallor. Click image to enlarge. |
Central Retinal Artery Occlusion (CRAO)
This condition is marked by a partial or complete obstruction of the CRA, resulting in retinal infarction. It’s most often caused by an embolic obstruction from atherosclerotic plaques in the carotid circulation or from cardiac valvular disease. These mechanisms are referred to as “non-arteritic” CRAO. In the case that CRAO occurs secondary to vasculitis, most commonly giant cell arteritis (GCA), it is referred to as “arteritic” CRAO.
The reported annual incidence of CRAO is 1.3 per 100,000 people.3 Although CRAO is described as an ophthalmic emergency, it should be reclassified as a medical emergency because of the associated risk of other life-threatening complications such as cerebrovascular accident (CVA), myocardial ischemia and cardiovascular death.4
Presentation. Patients with CRAO present with a unilateral, sudden, painless loss of vision. Timing of presentation is variable. In a series of 260 patients with CRAO, 23.1% sought medical attention on the same day or within one day of symptoms, 48.8% within one week, 74.6% within one month and the remaining within a few months.5
Examination. Perform a complete eye exam with attention to visual acuity, pupils (checking for an APD), intraocular pressure and the anterior segment (looking for signs of neovascularization). Visual acuity is count fingers or worse in 93.2% of non-arteritic cases.5 Present in 14.3% of cases is a cilioretinal artery, an arterial branch that originates from the lateral and medial posterior ciliary arteries and supplies part of the inner retina.5,6 In these cases, visual acuity is often better, ranging from 20/20 to count fingers or worse.5
Confrontation or formal visual field testing is optional, but patients with extensive central vision loss may have difficulty completing this task. Perform a dilated fundus exam. Findings include retinal whitening in the posterior pole (58%), a “cherry red spot” due to normal choroidal circulation in the fovea (90%), retinal arteriolar attenuation (32%), slow blood flow through the retinal arterioles (19%), optic disc edema (22%) and optic disc pallor (39%).7
Pathophysiology. An experimental study of rhesus monkeys with hypertension and atherosclerosis, in which the CRA was clamped at the site of entry to the ONH, demonstrated that CRAO lasting for 240 minutes results in massive irreversible retinal damage, seen both histologically and on electroretinogram.8 CRAO lasting 97 minutes showed minimal damage.8 Unlike many areas of the brain that have collateral circulation, the retinal ganglion cell layer is solely supplied by the CRA, with collateral capillaries present only around the optic disc.9 Thus, prolonged ischemia can have devastating consequences. Cilioretinal sparing protects the inner retina and results in preserved central visual function.
 |
|
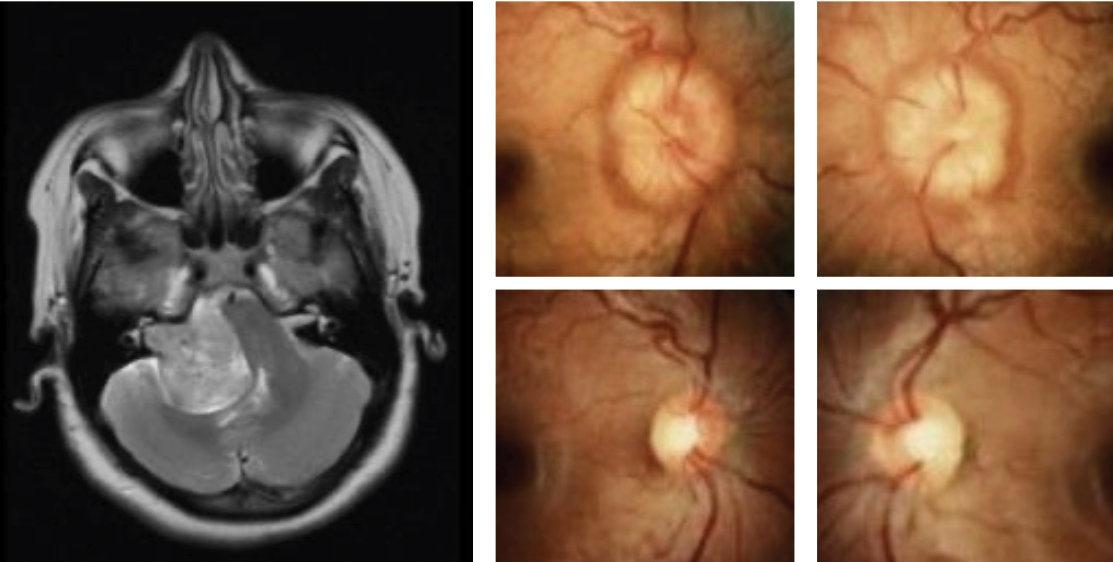
This pregnant female patient presented with pre-eclampsia. Following optimal treatment and improvement of blood pressure, resolution of hemorrhagic papilledema is noted with secondary disc pallor. On the bottom right is a typical visual field finding associated with papilledema. The degree of visual field suppression is secondary to the severity of disc involvement. Click image to enlarge. |
Workup. Tools to help with diagnosis include OCT and fluorescein angiography (FA). OCT at the time of acute CRAO may show retinal thickening and inner retinal hyperreflectivity.10 FA reveals absent or marked stasis of the retinal circulation.
CRAO should be referred emergently to a stroke center with access to on-call neurology, neuroradiology and cardiology.
Emboli can originate from atheromatous plaques in the internal carotid artery and cardiac valves.11 Occasionally, retinal emboli can be seen on fundus exam. One study concluded that retinal emboli are an unreliable indicator for hemodynamically significant carotid artery stenosis.12 Thus, perform carotid imaging on all patients in the form of ultrasound, computed tomography angiography (CTA) or magnetic resonance angiography (MRA).
Check the patient’s blood pressure. Perform electrocardiography (ECG) to evaluate for cardiac arrythmias such as atrial fibrillation that may predispose a patient to cardioembolic events. Conduct echocardiography to look for structural valvular lesions. Consult neurology as well.
Ask patients 55 and older about symptoms of GCA, including headache, myalgias, fever, unintended weight loss, jaw claudication and hip or shoulder pain. Palpate temporal artery pulses. If the history or exam is suggestive of GCA, order C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) and a platelet count. In a series of patients with biopsy-proven GCA, all those who presented with CRAO also had evidence of posterior ciliary artery circulatory abnormalities when FA was performed.13
Treatment. If clinical suspicion for GCA is present, initiate high-dose steroids while waiting on lab results to reduce the risk of further visual loss, involvement of the contralateral eye and other ischemic events (i.e., stroke and myocardial infarction). Confirm GCA with a temporal artery biopsy (TAB).
Unlike ischemic cerebral stroke, which has accepted treatment protocols, there are currently no evidence-based medical or surgical treatments that have been shown to affect the visual prognosis in CRAO.9 Interventions such as ocular massage, anterior chamber paracentesis, sublingual isosorbide dinitrate and hyperosmotic agents have been investigated but have shown minimal efficacy.14 Small case series and reports have documented improvement of visual acuity with prompt use of hyperbaric oxygen, but larger prospective randomized studies are needed to confirm whether this intervention affects prognosis.15,16
Management strategies should focus on preventing secondary vascular events.
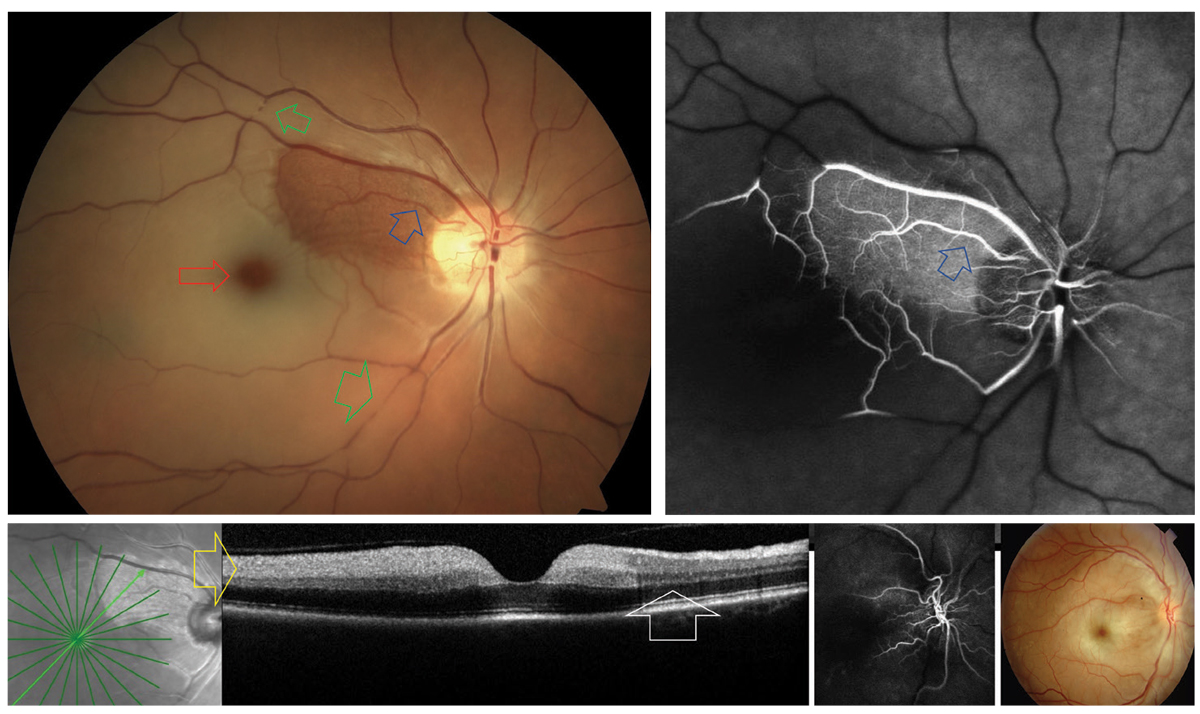 |
|
A cilioretinal artery (blue arrows) sparing CRAO. The areas of nonperfusion are noted to have retinal whitening with a prominent “cherry red spot” (red). Arterial plaques and segmentation “boxcarring” can be observed along the temporal vessels (green). On FA, significant vascular nonperfusion is noted outside the region supplied by the cilioretinal artery (blue). In the acute phase of CRAO, OCT typically reveals inner retinal hyperreflectivity (yellow). This is not present in the region spared by the cilioretinal artery (white). The upper right photo is an example of CRAO without cilioretinal sparing. Click image to enlarge. |
Non-Arteritic Anterior Ischemic Optic Neuropathy (NAION)
AION is a sudden cause of partial or complete unilateral, painless vision loss that can be broken down into arteritic (AAION) and NAION forms. NAION is defined as an idiopathic ischemic event affecting the anterior portion of the optic nerve. It is the most common optic neuropathy in the elderly population.17 Diabetes and systemic hypertension are common underlying causes.
The estimated annual incidence rate of NAION in patients over the age of 50 is 2.3 per 100,000 people.18 The mean age at presentation is 66, with a peak age range of 60 to 69.19 Systemic risk factors in patients older than 45 include hypertension, diabetes, ischemic heart disease, gastrointestinal ulcer and thyroid disease.20 Additional risk factors include nocturnal hypotension and obstructive sleep apnea (OSA).21-24 Small cup-to-disc ratio, seen in both the affected and fellow eyes, may play a role in the pathophysiology.25,26 The proposed mechanism is mechanical constriction of the optic nerve axons within the scleral canal.
Examination. The clinical diagnosis of NAION is made based on sudden, painless vision loss in the presence of optic nerve edema and a visual field defect.
Perform a comprehensive exam, including visual acuity, pupils (checking for an APD), color plates, confrontation and/or formal visual fields and dilated fundus exam. An APD is usually present unless there is bilateral symmetric eye involvement. Visual acuity is variable but not as drastic as the vision loss seen in AAION. Optic disc edema, either sectoral or general, is a prerequisite to diagnosing NAION. Examination of the fellow eye will reveal a small cup-to-disc ratio (“disc at risk”).25-27 Visual field abnormalities include inferior altitudinal defects, superior altitudinal defects, central scotoma, diffuse constriction and peripheral field loss.28
OCT and scanning laser polarimetry conducted on patients with NAION showed statistically significant sectoral retinal nerve fiber layer (RNFL) thinning corresponding to the affected hemifield on visual field testing.29 Interestingly, both diagnostic tests showed RNFL loss in the sectors corresponding to the unaffected hemifield as well.
The diagnosis of NAION is clinical. Lab tests are not indicated, and neither is neuroimaging nor referral to the ED. If the clinical picture or course is not consistent with a diagnosis of NAION, further workup may be warranted.
Treatment. Without intervention, 42.7% of patients with NAION experienced improvement of visual acuity in the affected eye by six months.30 Optic disc edema usually resolves by eight weeks.31 These patients have a 14.7% five-year risk of involvement of the fellow eye and should be counseled accordingly.32
The most important step in management is to differentiate NAION from AAION, as delayed diagnosis and inaccurate treatment can have devastating consequences.
Optic nerve sheath decompression may result in more vision loss.30 Visual recovery was seen in 42.7% of patients in the observation group at six months. Thus, this intervention has fallen out of favor.
Treatment with aspirin at the time of diagnosis does not affect visual acuity in the affected eye.33 There is conflicting evidence about whether aspirin serves as a protective agent for the fellow eye. One study reported that the initiation of aspirin at the time of diagnosis reduced the risk of second eye involvement at two years but that the protective effect was not present at five years.34 The incidence of second eye involvement in the Ischemic Optic Neuropathy Decompression Trial was not affected by the use of aspirin at five years. Despite the lack of evidence of long-term benefit with aspirin, many clinicians initiate this therapy due to the small side effect profile and the coexistence between NAION and other cardiovascular risk factors.
A recent randomized controlled trial showed no effect on visual acuity at six months with high-dose oral steroids.35 Greater reduction in optic disc edema on OCT was seen at one month in the treatment group, but the percentage change in RNFL in both the treatment and observation groups at six months was statistically similar.
Risk factor modification. Patients with NAION have a higher likelihood of experiencing myocardial infarctions and CVAs.36 Thus, a goal of management should be risk factor modification. If risk factors for OSA are present, suggest a sleep study.
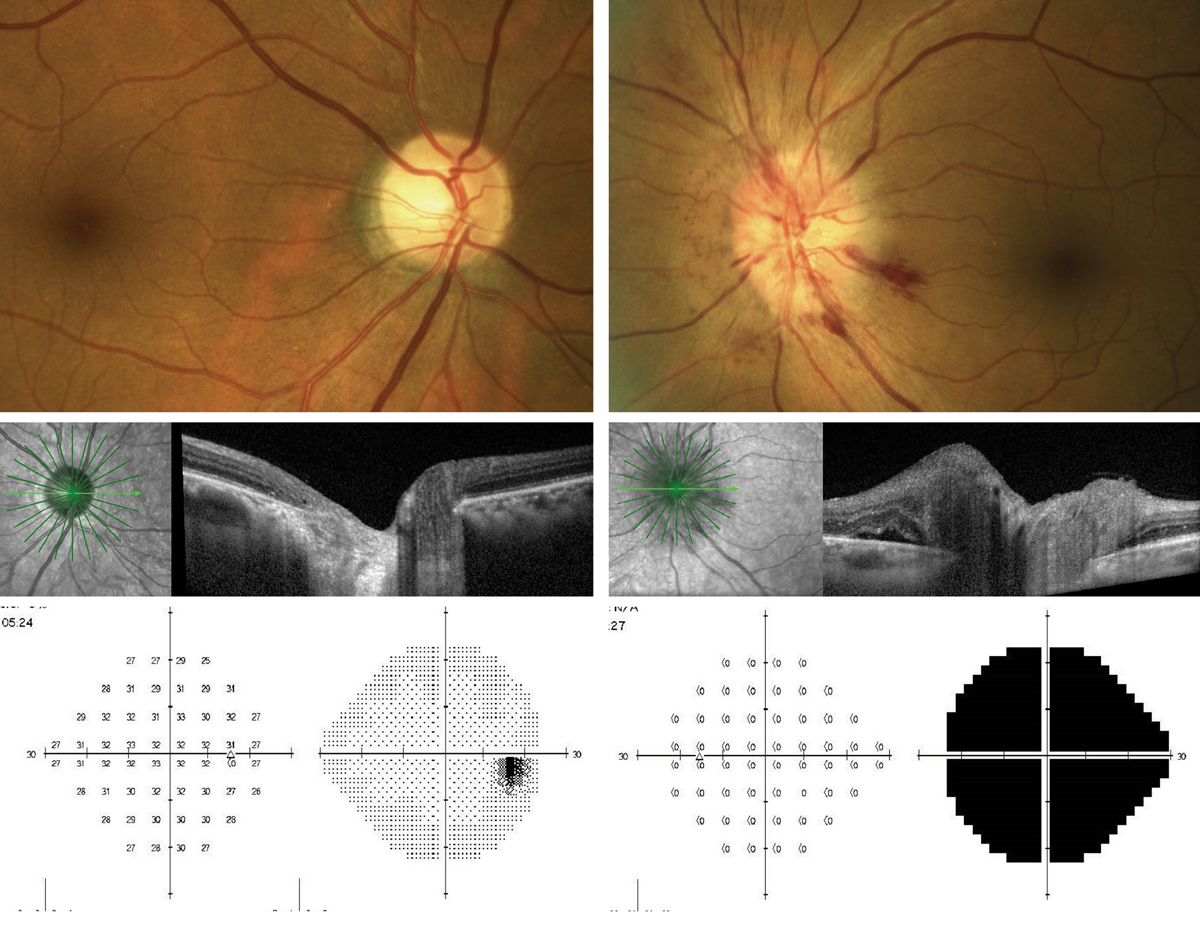 |
|
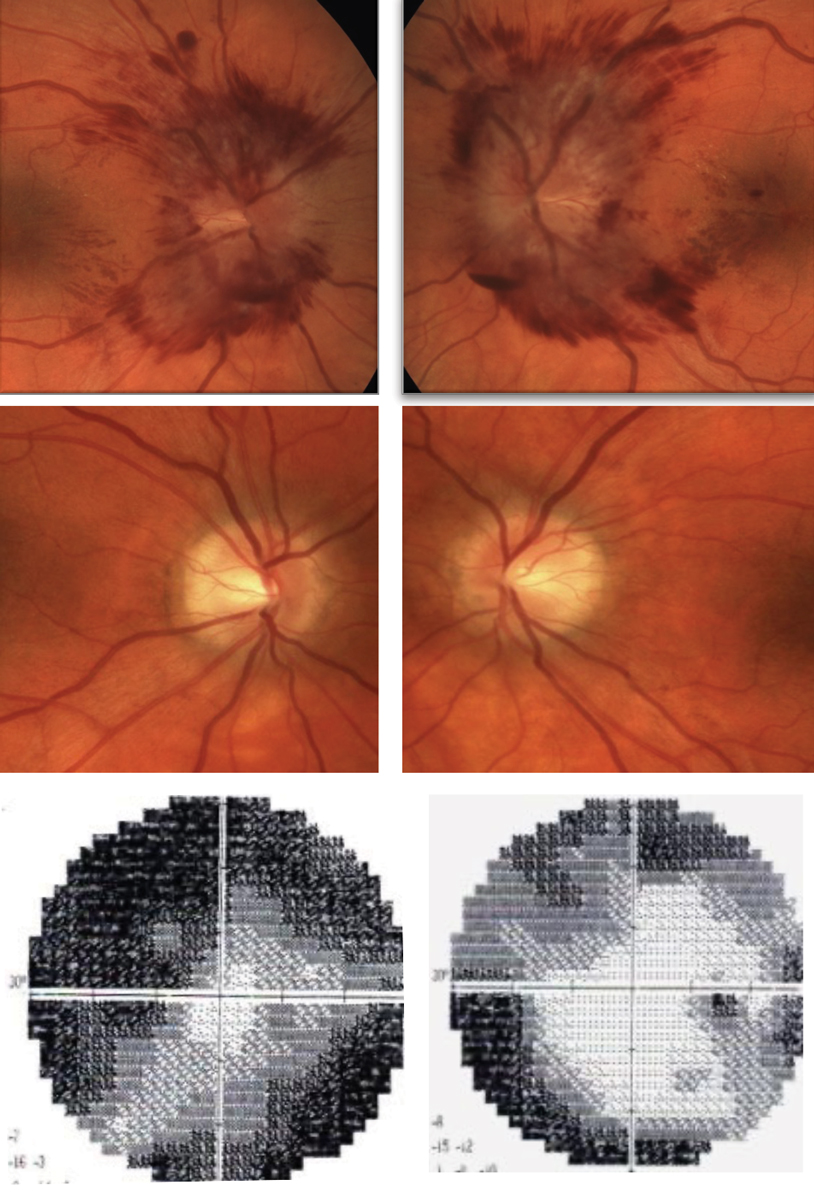
Presentation of AAION OS. Note the peripapillary hemorrhages and swelling of the optic nerve seen on OCT as compared with the normal right eye. The 24-2 threshold visual field OS shows significant depression of the left eye, while the right eye remains normal. Click image to enlarge. |
AAION
The arteritic form of AION is an ischemic event caused by GCA that affects the optic nerve. Patients with GCA are usually older, with a median age of 75. These patients can have temporal headaches, tenderness of the temples on the ipsilateral side and jaw or tongue claudication. The estimated annual incidence rate for AAION in patients over the age of 50 is 0.36 per 100,000 people.18
Presentation. AAION presents with acute, often severe, painless vision loss. Clinical features that should increase clinical suspicion for AAION include older age, pallid swelling (“chalky white appearance”) of the optic nerve and constitutional symptoms such as headache, scalp tenderness, weight loss, jaw claudication, anorexia, proximal muscle aches or weakness, fever, fatigue and malaise.37 Twenty-one percent of patients with GCA, however, can also present without systemic symptoms, a condition referred to as “occult GCA.” While no difference was seen in age or demographics, these patients tend to have lower ESR and CRP compared with patients who had classic symptoms.38
In contrast to NAION, visual acuity in AAION is much worse, ranging from hand motion to no light perception.37
In patients with biopsy-proven GCA presenting with AAION, the presence of chalky white disc edema was seen in 93% of eyes (59% during the first week, 20% during the second, 14% during the third, 5% during the fourth and 3% with unknown timing).13 Tender or non-pulsatile temporal arteries can be present on exam, but the absence of these findings does not rule out GCA.37
GCA can affect any medium- to large-sized blood vessel. Of the ocular arteries, the posterior ciliary ones, which supply the choroid and the anterior portion of the optic nerve, are the most affected.37 As the retina and the optic nerve are very sensitive to ischemic damage, the visual prognosis after an ischemic event such as AAION is very poor.
Differential diagnosis. The clinical diagnosis of AAION is made based on degree of vision loss, timing of symptoms, appearance of the optic disc and a concurrent diagnosis of GCA.
The American College of Rheumatology lists five criteria as the gold standard for diagnosis of GCA:39
- age ≥50
- new-onset of localized headache
- temporal artery tenderness or decreased temporal artery pulse
- elevated ESR ≥50mm/hour
- positive TAB
The absence of elevated inflammatory markers doesn’t rule out GCA.40-42 Elevated CRP and ESR have a sensitivity of 86.9% and 84.1%, respectively, for predicting a positive TAB.42
Treatment. AAION is a medical emergency and thus should be sent to the ED for workup and initiation of high-dose corticosteroids. If there is a logistical delay in getting the patient to the ED, initiate high-dose oral steroids in the office. Ideally, calling the ED ahead of time should reduce the likelihood of the patient being stuck in triage or the waiting room and not receiving timely treatment.
Do not delay treatment for lab confirmation. Treatment includes intravenous methylprednisolone 250mg every six hours for three to five days.43 Some advocate for the use of concurrent aspirin as well. Steroid therapy is then transitioned to oral prednisone 1mg/kg/day until ESR and CRP normalize.
Labs include ESR, CRP and a complete blood count. In a comparison of patients with AAION vs. NAION, those with AAION had higher ESR, CRP, platelet counts and white blood cell counts and lower hemoglobin and hematocrit levels.44
Once high-dose steroids have been initiated, perform TAB within seven to 10 days to avoid a false negative diagnosis. A positive TAB is the gold standard for diagnosis of GCA. Due to the segmental nature of the granulomatous lesions, a biopsy of at least 2cm in length should be performed. TAB has a sensitivity of 87.1%.45 A positive biopsy has a 100% specificity.43
Long-term immunomodulation therapy is best performed by a rheumatologist.
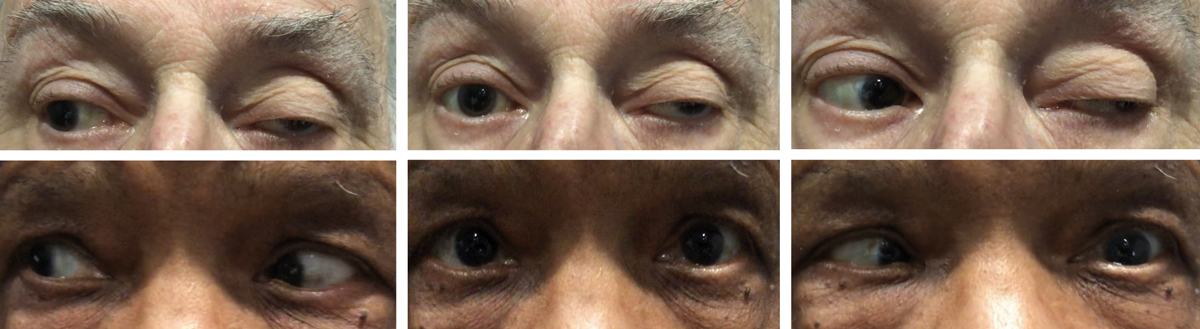 |
|
Cranial nerve III palsy with exotropia, hypotropia and ptosis OS (top). There is restriction of left adduction, supraduction and infraduction. Note, both pupils are pharmacologically dilated. Isolated cranial nerve VI palsy OS (bottom). Note the subtle left esotropia in primary gaze with complete loss of abduction and normal adduction. Click image to enlarge. |
Sudden-onset Diplopia
This condition has a broad differential diagnosis and can be subcategorized into monocular and binocular forms. Causes of monocular diplopia include dry eye, cataract and epiretinal membrane. Monocular diplopia is never caused by a neurologic abnormality. It can be elicited on exam by resolution of symptoms with monocular occlusion or pinhole testing.
In contrast, binocular diplopia is caused by ocular misalignment and warrants a neurologic review of systems, physical exam and, in many cases, neuroimaging. Clarify timing of symptoms, if symptoms fluctuate during the day and whether there is eyelid or pupil involvement. Question patients with binocular diplopia over the age of 50 about symptoms of GCA, as GCA can present as an ischemic cranial nerve palsy. Use alternate cover testing to determine the amount and direction of ocular misalignment, noting whether the misalignment is comitant or incomitant.
In comitant misalignment, the deviation remains the same in all gazes. In incomitant misalignment, the deviation depends on position of gaze. Comitant misalignment is usually secondary to decompensated strabismus and does not require additional neurologic workup. In contrast, incomitant misalignment warrants additional history and workup.46 The clinician should pay careful attention to lid position, pupil symmetry and reaction to light.
Isolated cranial neuropathies. These can occur secondary to compression, ischemia, trauma and inflammation. There is controversy in the management of an isolated cranial nerve palsy in the absence of other neurologic deficits. One study found that in a cohort of 93 patients older than 50 with acute isolated mononeuropathies including cranial nerve III, IV and VI palsies without other neurologic dysfunction, 4.3% were found to have lesions on MRI and only 1.1% were thought to have a structural lesion deemed to be causative of the cranial nerve palsy.47 The researchers concluded that neuroimaging may not be warranted in patients with isolated cranial nerve palsies without evidence of other neurologic deficits. A similar prospective study found a higher prevalence of structural lesions in 14% of patients with isolated cranial neuropathies.48 This team concluded that neuroimaging should be included in the workup.
Ischemic cranial nerve palsies improve over six months, whereas compressive cranial nerve palsies do not self-resolve and may present with other neurologic deficits. If neuroimaging is not initially obtained, monitor patients closely to ensure resolution of the cranial neuropathy. If resolution is not seen or if other neurologic abnormalities are seen, obtain neuroimaging.
Cranial nerve III palsy. The third nerve (oculomotor) has several branches. The superior branch innervates the levator palpebrae superioris (LPS) and the superior rectus. The inferior branch innervates the medial rectus, inferior rectus and inferior oblique muscles. The superior rectus is innervated by the contralateral cranial nerve III nucleus. The LPS muscles are innervated by a joined single nucleus. Thus, an oculomotor nuclear lesion results in bilateral ptosis.
Compression of the third nerve by an aneurysm causes pupillary dilation due to involvement of the parasympathetic fibers. Compressive third nerve palsies can present without ptosis, mydriasis or extraocular muscle deficits. This phenomenon is referred to as “incomplete.”
If complete paresis is seen of all the extraocular muscles innervated by cranial nerve III but the pupil is normal (i.e., pupil-sparing), then an aneurysm can be ruled out. Some recommend patients be closely observed without neuroimaging in these cases. Others recommend conducting neuroimaging of all cranial nerve III palsies.
An unrecognized cerebral aneurysm can be a life-threatening emergency. Thus, carefully examine any patient who presents with diplopia and ptosis and keep cranial nerve III palsy high on the differential diagnosis. Urgent referral to a regional medical center with on-call neurologists, neuroradiologists and neurosurgeons is ideal if there is any concern for a compressive third nerve palsy. CT and CTA of the brain are included in the workup.
Cranial nerve IV palsy. The fourth nerve (trochlear) innervates the contralateral superior oblique muscle. The fourth nerve has the longest intracranial course of all cranial nerves and is thus most susceptible to trauma.
Patients with cranial nerve IV palsy present with ipsilateral hypertropia. The three-step test is used to make the diagnosis. Hypertropia increases in contralateral gaze and ipsilateral head tilt. One of the actions of the superior oblique is incyclotorsion; thus, patients with trochlear nerve palsies will have some degree of excyclotorsion.
Longstanding trochlear nerve palsies are differentiated from acute palsies based on the presence of old photos showing head tilt, facial asymmetry and increased convergence amplitudes.
Cranial nerve VI palsy. The sixth nerve (abducens) innervates the ipsilateral lateral rectus muscle. Thus, an abducens palsy presents with esotropia and limited abduction on the side ipsilateral to the lesion.
There is controversy in the management of an isolated sixth nerve palsy. Some advocate for neuroimaging, while others advocate for close observation if microvascular risk factors such as hypertension and diabetes are present and there are no other neurologic deficits. The natural course of a microvascular cranial neuropathy is spontaneous improvement over six months. If clinical improvement is not seen during this time or other cranial nerves are involved, then neuroimaging is indicated.
Multiple cranial neuropathies. When multiple cranial nerve abnormalities are localized on exam, urgent referral to the ED for imaging of the brain and orbit is indicated, specifically to evaluate the orbital apex, cavernous sinus and brainstem. Ideally, refer patients to a regional medical center with access to an on-call neurologist, neuroradiologist and neurosurgeon.
Other motility deficits. When multiple extraocular motility deficits and/or eyelid ptosis is seen that does not fit the pattern of a single or multiple cranial nerve palsies, consider myasthenia gravis, thyroid eye disease, Miller Fisher syndrome and skew deviation. Pupil involvement is not seen in myasthenia gravis. Outpatient referral to a neurologist or neuro-ophthalmologist can assist with workup.
 |
|
This patient developed diplopia after a car accident. He has a cranial nerve IV palsy with an up and out deviation OD shown in primary gaze, which is exaggerated by the traumatic scar on the lower lid of the same eye. Click image to enlarge. |
Other Presentations of Acute-onset Vision Loss
Patients can present with vision loss (unilateral or bilateral) without any significant physical ocular findings to support their claims. The following are a few scenarios and discussion of how to manage each:
Scenario one. A 70-year-old African American male with history of hypertension and diabetes presents with complaints of significant vision loss OD. Visual acuities are 20/30 OD and 20/20 OS. His history is positive for cataract surgery over 10 years ago, and there are no anterior or posterior segment findings to support his complaint. Visual fields reveal a right homonymous hemianopsia consistent with a posterior CVA.
Refer this patient to a stroke center for urgent CT of the brain to rule out hemorrhagic stroke, followed by MRI of the brain and either CTA or MRA to assess for vascular compromise. Cardiac workup including ECG and echocardiography are included in the stroke evaluation. If an ischemic stroke is recognized acutely and the patient is within the window for treatment with tissue plasminogen activator, then the neurologic deficits can be restored. However, oftentimes presentation to a medical provider is delayed. Goals of therapy are risk factor identification and modification. Outpatient referral is inappropriate in this context, even if symptoms are subacute.
Scenario two. A 34-year-old white female presents with sudden decrease of vision OS. She has no remarkable medical or ocular history. Her visual acuities are 20/20 OD and 20/60 OS. Pupils are equally reactive; however, she has an APD OS. She reports pain with eye movement. Otherwise, there are no remarkable anterior or posterior ocular findings to support the vision loss or the APD. Visual field testing shows a statistically remarkable overall depression of the left eye. Suspicion of retrobulbar optic neuritis is indicated.
Refer this patient to the ED for an urgent MRI of the brain and orbits with gadolinium and fat suppression to evaluate for optic neuritis. Per the Optic Neuritis Treatment Trial, the risk of recurrence of optic neuritis was lower in patients who received IV steroids as opposed to oral steroids.49 MRI is also indicated to evaluate for periventricular white matter lesions, which if seen would result in a diagnosis of multiple sclerosis (MS). Even in the absence of white matter lesions, counsel the patient on the long-term risk of developing MS. Other conditions that can have similar presentations include optic perineuritis (usually seen in infectious or inflammatory conditions) and neuromyelitis optica.
Scenario three. A 25-year-old male presents with sudden-onset vision loss OD and 20/200 visual acuity. However, all his examination findings are normal. There is no APD or signs of dry eye, keratoconus, cataract, inflammatory disease, retinal disease or macular disease. His visual field testing is inconclusive.
In the absence of an APD and no historical or exam findings suggestive of a neurologic abnormality, referral to the ED is not warranted and, without a working diagnosis or planned workup, can result in an unfocused, expensive shotgun workup that is usually of low diagnostic yield. Perform refraction to ensure vision is at its best-corrected potential. Establish close follow-up to ensure reproducibility of exam findings. Perform other tests such as tangent screening to rule out malingering. Factitious vision loss is a diagnosis of exclusion.
 |
|
Visual field defects in patients with sudden-onset vision loss with no significant anatomic ocular findings. On top, a patient is suffering from a right homonymous hemianopia caused by a left hemispheric CVA. These patients often perceive loss in the eye on the same side as their hemianopsia. In this case, the patient perceived loss of vision OD. Below, a patient is suffering from retrobulbar optic neuritis OD. These patients present with varying degrees of overall vision and visual acuity loss, often significantly reduced. Their examination is usually remarkable for pain with eye movement, an APD and red-colored desaturation. MRI of the brain and orbits with contrast and fat suppression is indicated to confirm optic nerve enhancement and to assess for periventricular white matter changes, indicative of MS. Click image to enlarge. |
Referral to the ED
Most of the conditions discussed here can have devastating neurologic consequences if not evaluated in an urgent manner. Ideally, refer these patients to a regional medical center with access to on-call neurology, neuroradiology and board-certified emergency medicine doctors who can offer the highest level of care.
Provide either a copy of your exam notes or a letter documenting the patient’s ocular exam findings, suspected diagnosis and recommended workup. If possible, call the ED and speak to the appropriate physician about your patient.
Ensure close follow-up is established post-hospitalization or arrange the appropriate follow-up with the appropriate subspecialist.
Dr. Mohammad Rafieetary practices at the Charles Retina Institute in Germantown, TN. He is a speaker and consultant for Heidelberg Engineering, Notal Vision, Optos, Regeneron Pharmaceutical, Spark Therapeutics and Apellis Therapeutics. Dr. Salar Rafieetary also practices at the Charles Retina Institue. Dr. Briggs works at the University of Alabama Department of Emergency Medicine in Mobile, AL. Drs. Salar Rafieetary and Briggs have no financial interests to disclose.1. Rigi M, Almarzouqi SJ, Morgan ML, Lee AG. Papilledema: epidemiology, etiology, and clinical management. Eye Brain. 2015;7:47-57. 2. Crum OM, Kilgore KP, Sharma R, et al. Etiology of papilledema in patients in the eye clinic setting. JAMA Netw Open. 2020;3(6):e206625. 3. Leavitt JA, Larson TA, Hodge DO, Gullerud RE. The incidence of central retinal artery occlusion in Olmsted County, Minnesota. Am J Ophthalmol. 2011;152(5):820-3. 4. Dattilo M, Biousse V, Newman NJ. Update on the management of central retinal artery occlusion. Neurol Clin. 2017;35(1):83-100. 5. Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol. 2005;140(3):376-91. 6. Michalinos A, Zogana S, Kotsiomitis E, et al. Anatomy of the ophthalmic artery: a review concerning its modern surgical and clinical applications. Anat Res Int. 2015;2015:591961. 7. Hayreh SS, Zimmerman MB. Fundus changes in central retinal artery occlusion. Retina. 2007;27(3):276-89. 8. Hayreh SS, Zimmerman MB, Kimura A, Sanon A. Central retinal artery occlusion. Retinal survival time. Exp Eye Res. 2004;78(3):723-36. 9. Chronopoulos A, Schutz JS. Central retinal artery occlusion—a new, provisional treatment approach. Surv Ophthalmol. 2019;64(4):443-51. 10. Shah P, Schwartz SG, Flynn HW. Multimodal images of acute central retinal artery occlusion. Case Rep Ophthalmol Med. 2017;2017:5151972. 11. Recchia FM, Brown GC. Systemic disorders associated with retinal vascular occlusion. Curr Opin Ophthalmol. 2000;11(6):462-7. 12. Sharma S, Brown GC, Pater JL, Cruess AF. Does a visible retinal embolus increase the likelihood of hemodynamically significant carotid artery stenosis in patients with acute retinal arterial occlusion? Arch Ophthalmol. 1998;116(12):1602-6. 13. Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125(4):509-20. 14. Rumelt S, Dorenboim Y, Rehany U. Aggressive systematic treatment for central retinal artery occlusion. Am J Ophthalmol. 1999;128(6):733-8. 15. Beiran I, Goldenberg I, Adir Y, et al. Early hyperbaric oxygen therapy for retinal artery occlusion. Eur J Ophthalmol. 2001;11(4):345-50. 16. Kim YS, Nam MS, Park EJ, et al. The effect of adjunctive hyperbaric oxygen therapy in patients with central retinal artery occlusion. Undersea Hyperb Med. 2020;47(1):57-64. 17. Hattenhauer MG, Leavitt JA, Hodge DO, et al. Incidence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1997;123(1):103-7. 18. Johnson LN, Arnold AC. Incidence of nonarteritic and arteritic anterior ischemic optic neuropathy. Population-based study in the state of Missouri and Los Angeles County, California. J Neuroophthalmol. 1994;14(1):38-44. 19. Characteristics of patients with nonarteritic anterior ischemic optic neuropathy eligible for the Ischemic Optic Neuropathy Decompression Trial. Arch Ophthalmol. 1996;114(11):1366-74. 20. Hayreh SS, Joos KM, Podhajsky PA, Long CR. Systemic diseases associated with nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1994;118(6):766-80. 21. Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994;117(5):603-24. 22. Mojon DS, Hedges TR, Ehrenberg B, et al. Association between sleep apnea syndrome and nonarteritic anterior ischemic optic neuropathy. Arch Ophthalmol. 2002;120(5):601-5. 23. Bilgin G, Koban Y, Arnold AC. Nonarteritic anterior ischemic optic neuropathy and obstructive sleep apnea. J Neuroophthalmol. 2013;33(3):232-4. 24. Wu Y, Zhou LM, Lou H, et al. The association between obstructive sleep apnea and nonarteritic anterior ischemic optic neuropathy: a systematic review and meta-analysis. Curr Eye Res. 2016;41(7):987-92. 25. Beck RW, Savino PJ, Repka MX, et al. Optic disc structure in anterior ischemic optic neuropathy. Ophthalmology. 1984;91(11):1334-7. 26. Feit RH, Tomsak RL, Ellenberger C. Structural factors in the pathogenesis of ischemic optic neuropathy. Am J Ophthalmol. 1984;98(1):105-8. 27. Doro S, Lessell S. Cup-disc ratio and ischemic optic neuropathy. Arch Ophthalmol. 1985;103(8):1143-4. 28. Gerling J, Meyer JH, Kommerell G. Visual field defects in optic neuritis and anterior ischemic optic neuropathy: distinctive features. Graefes Arch Clin Exp Ophthalmol. 1998;236(3):188-92. 29. Deleón-Ortega J, Carroll KE, Arthur SN, Girkin CA. Correlations between retinal nerve fiber layer and visual field in eyes with nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 2007;143(2):288-94. 30. Optic nerve decompression surgery for nonarteritic anterior ischemic optic neuropathy (NAION) is not effective and may be harmful. The Ischemic Optic Neuropathy Decompression Trial Research Group. JAMA. 1995;273(8):625-32. 31. Hayreh SS, Zimmerman MB. Optic disc edema in non-arteritic anterior ischemic optic neuropathy. Graefes Arch Clin Exp Ophthalmol. 2007;245(8):1107-21. 32. Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow-up study. Am J Ophthalmol. 2002;134(3):317-28. 33. Botelho PJ, Johnson LN, Arnold AC. The effect of aspirin on the visual outcome of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1996;121(4):450-1. 34. Beck RW, Hayreh SS, Podhajsky PA, et al. Aspirin therapy in nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1997;123(2):212-7. 35. Saxena R, Singh D, Sharma M, et al. Steroids versus no steroids in nonarteritic anterior ischemic optic neuropathy: a randomized controlled trial. Ophthalmology. 2018;125(10):1623-7. 36. Guyer DR, Miller NR, Auer CL, Fine SL. The risk of cerebrovascular and cardiovascular disease in patients with anterior ischemic optic neuropathy. Arch Ophthalmol. 1985;103(8):1136-42. 37. Hayreh SS. Ophthalmic features of giant cell arteritis. Baillieres Clin Rheumatol. 1991;5(3):431-59. 38. Hayreh SS, Podhajsky PA, Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998;125(4):521-6. 39. Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122-8. 40. Yoeruek E, Szurman P, Tatar O, et al. Anterior ischemic optic neuropathy due to giant cell arteritis with normal inflammatory markers. Graefes Arch Clin Exp Ophthalmol. 2008;246(6):913-5. 41. Poole TR, Graham EM, Lucas SB. Giant cell arteritis with a normal ESR and CRP. Eye (Lond). 2003;17(1):92-3. 42. Kermani TA, Schmidt J, Crowson CS, et al. Utility of erythrocyte sedimentation rate and C-reactive protein for the diagnosis of giant cell arteritis. Semin Arthritis Rheum. 2012;41(6):866-71. 43. Scheurer RA, Harrison AR, Lee MS. Treatment of vision loss in giant cell arteritis. Curr Treat Options Neurol. 2012;14(1):84-92. 44. Costello F, Zimmerman MB, Podhajsky PA, Hayreh SS. Role of thrombocytosis in diagnosis of giant cell arteritis and differentiation of arteritic from non-arteritic anterior ischemic optic neuropathy. Eur J Ophthalmol. 2004;14(3):245-57. 45. Niederkohr RD, Levin LA. A Bayesian analysis of the true sensitivity of a temporal artery biopsy. Invest Ophthalmol Vis Sci. 2007;48(2):675-80. 46. Margolin E. Approach to patient with diplopia. J Neurol Sci. 2020;417:117055. 47. Murchison AP, Gilbert ME, Savino PJ. Neuroimaging and acute ocular motor mononeuropathies: a prospective study. Arch Ophthalmol. 2011;129(3):301-5. 48. Chou KL, Galetta SL, Liu GT, et al. Acute ocular motor mononeuropathies: prospective study of the roles of neuroimaging and clinical assessment. J Neurol Sci. 2004;219(1-2):35-9. 49. Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326(9):581-8. |
