 |
A 58-year-old Black female presented for a comprehensive ocular examination with a chief complaint of poor vision OD after being a passenger in an auto accident, during which she was struck in the face with an airbag. The patient denied any additional ocular history and reported a medical history of hypothyroidism, currently well-controlled with medication. The patient denied having any allergies to medications or environment. She is currently taking levothyroxine, lisinopril/hydrochlorothiazide, allopurinol, sertraline and ranitidine.
Diagnostic Data
Her best-corrected entering visual acuities were 20/20 OD and 20/30 OS at distance and near OU with no improvement upon pinhole. Her external examination was remarkable for superior constriction upon confrontation fields OD with a grade 1 afferent pupillary defect OD. Biomicroscopic evaluation of the anterior segment was unremarkable, with normal Goldmann applanation tonometry pressures of 17mm Hg OU. The pertinent retinal findings are demonstrated in the photograph.
Additional testing included baseline photodocumentation, baseline optical coherence tomography and baseline automated perimetry. Since “significant mechanism of injury” trauma, as in this case, can produce anatomical damage that can induce secondary open-angle glaucoma, gonioscopy was also completed, uncovering an angle open to the ciliary body without angle recession.
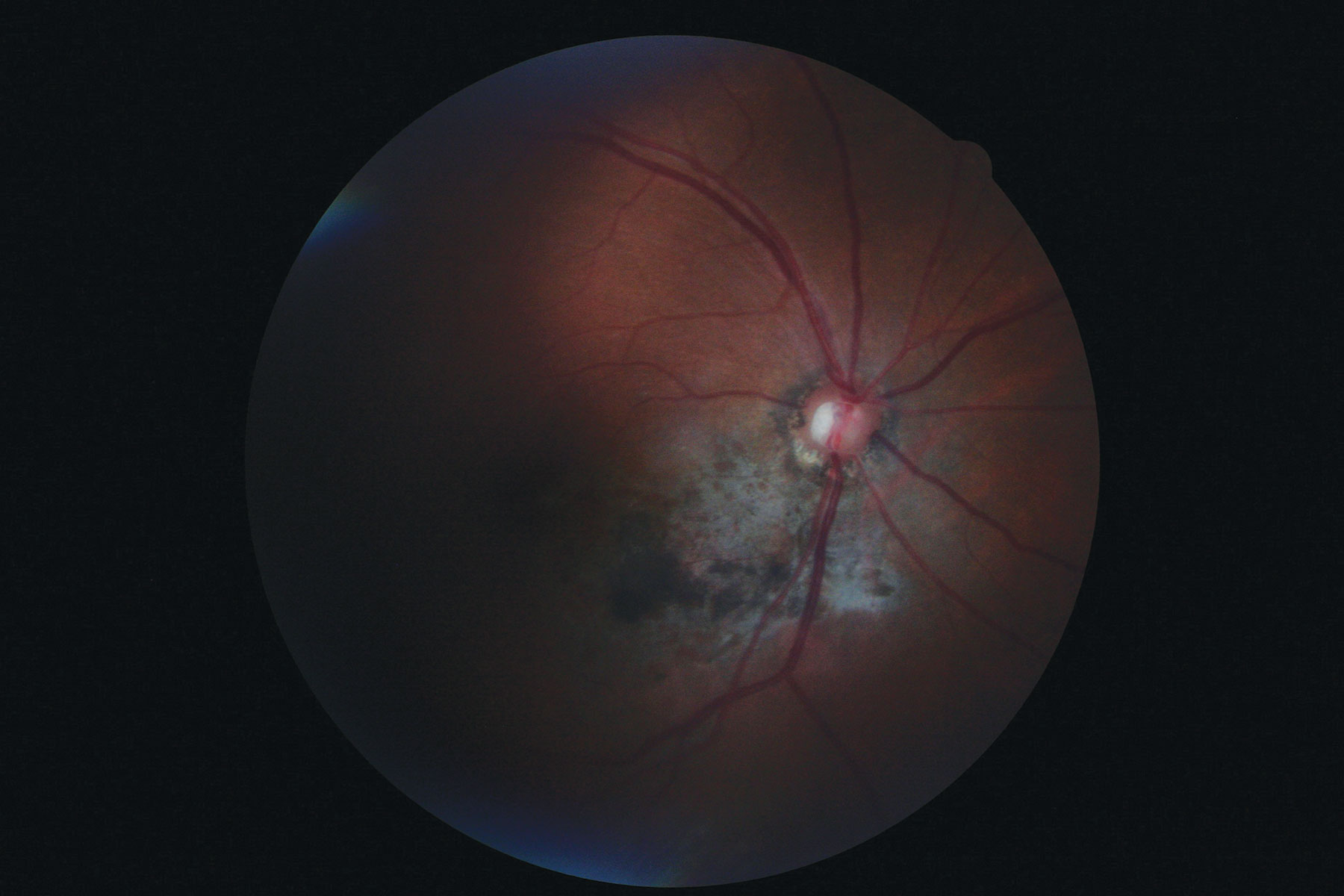 |
| Dilated fundus exam of the patient revealed the findings shown here. In what way might they relate to the incident (an auto accident) she described upon presentation? Click image to enlarge. |
Diagnosis
The diagnosis in this issue is traumatic chorioretinal trauma with retinal pigment epithelial (RPE) clumping, choroidal rupture without choroidal neovascularization and traumatic optic neuropathy resulting in secondary nerve fiber layer compromise and sectoral visual field loss.
Blunt trauma to the eye and adnexa can produce a number of acute and chronic ocular sequelae, ranging from adnexal abrasions, lacerations with or without corneal injury, hyphema and broken orbital bones (with or without extraocular muscle entrapment) to vitreous hemorrhage, retinal detachment, choroidal rupture, commotio retinae and traumatic optic neuropathy.1-21
The mechanism of blunt ocular coup and contrecoup trauma is well described in the literature.1-59
One of the secondary complications of blunt ocular trauma is commotio retinae, also called Berlin’s edema. It presents upon funduscopy as an area of milky white intraretinal edema following blunt ocular trauma.1-14 The condition does not occur spontaneously. While the entity is painless, its symptoms are associated with the location and extent of the retinal bruising along with the collateral consequences of the blunt trauma.1-18 Any amount of intraretinal fluid located centrally can cause patients to observe metamorphopsia or reduced visual acuity without decreased red or brightness saturation. The ocular signs of blunt trauma can range from mild diffuse injection, chemosis, subconjunctival hemorrhage, photophobia and pain upon ocular motility to orbital fracture, iridodialysis, lens luxation, traumatic iritis, vitreous hemorrhage and globe rupture.2-13 While specific data for ocular injuries have fluctuated over the last 10 years, one thing has remained constant: injury rates are consistently highest among males of all ages.6-11
The classic work of Sipperly, Quigley and Gass demonstrated the predominant retinal abnormality in commotio retinae (Berlin’s edema) was disruption of the photoreceptor outer segments caused by hydraulic ocular distention induced by blunt force trauma.1,15 This mechanism alters the hydrodynamics of the intraretinal and choroidal vascular systems, creating fluid leakage. The condition is not merely an accumulation extracellular fluid, as originally postulated by Berlin.1-4,14,15,28-31 Extensive work by researchers has documented in both monkey and pig models that the predominant pathophysiology is destruction of the vulnerable and delicate photoreceptor outer segments along with the concomitant imbibition of multiple intraretinal cellular elements (RPE, axons of the photoreceptor cells, Müller’s cells and selected ganglion cells) with intracellular fluid.1-4,14,15,28-31
The capacity for visual disability depends upon the volume and location of permanently lost photoreceptors. Interestingly, the pigment epithelium’s response to traumatic photoreceptor damage seems to be similar to that observed in experimental retinal detachment and light-induced retinal damage.13,14 Recent in-vivo investigations using OCT confirms what was postulated by previous histological studies: commotio retinae is caused by and consists of a disruption and fragmentation of the photoreceptor outer segments with resultant fluid incursion and collateral damage to retinal elements.1-4,14-17 The location of the commotio injury is traditionally in the contrecoup position or 180 degrees opposite the site of the impact.1,18
Researchers have attempted to use spectral domain OCT imaging to create a grading scale for macular commotio retinae based upon four distinct photoreceptor morphologic features that have been consistently observed:4
Grade 1—an increase in inner segment-outer segment (IS-OS) junction reflectivity with the disappearance of the thin hyporeflective optical space.
Grade 2—cone outer segment tip (COST) defects only.
Grade 3—COST and IS-OS junction defects.
Grade 4—COST defects, IS-OS junction and external limiting membrane (ELM) defects.
The group was also able to conclude that eyes with higher grades at baseline had significantly worse visual and anatomic outcomes.4
Cystoid macular edema (CME) is a classical complication of ocular inflammation and is possible comorbidity of commotio retinae.21 This syndrome was described by Irvine in 1953,;however, the pathogenesis of this condition remains unclear.12 Cystoid macular edema can result either from an insult to the inner or outer blood-ocular barrier.12,21
Examination and Care
There is no treatment for commotio retinae.12-14,33-37 The ocular management consists of attending to the accompanying damage (facial or orbital fractures, tissue lacerations, corneal abrasion, iritis, subconjunctival hemorrhage) and emotionally supporting the patient when the vision is reduced. Visual acuity monitoring, Amsler grid testing, interferometry and SD-OCT measurements can provide comparative data indicating the progress of recovery.1-4,14-17 There is no topical, oral or surgical solution proper.
Patients can be counseled that in the absence of visual axis pathology (cornea, lens, vitreous, macula), visual recovery often returns to pre-injury levels.16-18 In the event it does not, the majority of incompletely resolved cases return to levels better than 20/30.18 Unfortunately, responsible discussions should include the possibility that acuity will remain disabled. Complications such as premature cataractogenesis and the symptoms associated with epiretinal membrane formation, posterior vitreous detachment and macular hole formation should also be mentioned.1,3,5,6,11-13,33-37
Choroidal rupture occurs when the forces of blunt trauma create sudden tensile stress on the tissue layers in the eye wall induced by sudden deformation of the globe.38 This creates a break in Bruch's membrane and underlying choriocapillaris with preservation of the overlying retina and retinal layers.38,39 A traumatic choroidal rupture is similar, but includes the simultaneous rupture of both the choroid and overlying retina and a retraction of these tissues to reveal underlying sclera.38 Here, subretinal hemorrhage is common and injury to subretinal and retinal tissue is common.38,39 Approximately 5% to 10% of patient's presenting with blunt ocular trauma will develop a choroidal rupture.39 The early presence of deep subretinal hemorrhages should raise the suspicions of choroidal rupture.38,39 If the affected region is local to the macula, visual acuity may be profoundly and permanently affected. Any time this occurs, the secondary formation of choroidal neovascularization is plausible and will require permanent monitoring. In severe cases of blunt ocular trauma, retrobulbar hemorrhage is possible.38,39
When the optic nerve is compromised by a severe blow, traumatic optic neuropathy ensues. Patients with traumatic optic neuropathy typically experience sudden, severe, unilateral vision loss following blunt injury to the head or face.1-3 The condition may manifest immediately or within hours or days following the trauma.4 The vision loss may be insidious, with some patients being unaware of any visual deficit until it is detected by routine examination.
The epidemiology of traumatic optic neuropathy is non-specific; however, it can be extrapolated as following trends consistent with those individuals most likely to sustain both eye injuries and general blunt ocular trauma.5-10,33-45 The patient set that is most prone to both ocular injury and ocular blunt trauma includes young males (an average of 4:1 over females), in their teens or twenties, participating in outdoor or work-related activities during climate seasonal weather.5-10 Women who sustain serious ocular injury tend to be involved in activities around the home.7 The history often includes a blow to the head severe enough to induce loss of consciousness or high speed penetration of the globe by foreign material.1-10
Examination of these patients reveals variably reduced acuity and/or visual field defects, which may be central, paracentral, arcuate or altitudinal in nature.1-10 An afferent pupillary defect is characteristic and telltale for severity with varying degrees of dyschromatopsia noted.1-11 Ophthalmoscopic evaluation will vary greatly depending on the nature and severity of the injury. Initially, practitioners may note a completely normal fundus without any signs of disc edema, ischemia or other abnormalities.12,13 In other cases, a grossly edematous optic nerve head, vitreous hemorrhage, venous congestion or retinal edema may be seen.1-12 In the vast majority of cases, optic disc pallor ensues within several weeks to months after the injury.1-4,12-14,33-45
Traumatic optic neuropathy results from injury sustained during trauma to the orbital rim or frontal area.12-14,27-45 In adults, the etiology is typically a bicycle or motor vehicle accident, but may also include physical assault, falls, sports-related injuries or (rarely) orbital surgery.1-11,27-45 There are a variety of ways in which the optic nerve can be damaged. In some cases the pathophysiology may be multifactorial. Mechanisms include transection or avulsion of the nerve, hematoma of the nerve sheath, optic nerve compression secondary to bony fracture of the orbital apex or penetrating orbital foreign body.1-12 Most commonly, however, concussive shock waves are implicated; transmission of these forces to the bones and meninges of the orbit results in contusion of the intracanalicular optic nerve.5-14 Subsequently, the axons and microvasculature are compromised by ischemia secondary to reactive edema, as well as the generalized compressive forces.14 In rare instances, the neuropathy may develop months after the initial trauma, a consequence of scarring within the optic canal that leads to secondary nerve compression.13,44-59
Trauma that precipitates an optic nerve injury almost always results in retinal ganglion cell axon damage.14 Neurotrophin deprivation secondary to direct shear, edema, bleeding or other neurochemotactic factors induce death of the retinal ganglion cells (RGCs).14 These cells give rise to the axons that comprise the optic nerve. Externally released agents that block mitochondrial electron transport have been used in the laboratory to show that superoxide radicals generated in the mitochondrial electron transport chain also induce cell death after axonal injury.14
High-resolution imaging of the head is critical in any case of blunt trauma to ensure that no life-threatening intracranial damage has been sustained.15 Particular attention should be given to the orbital apex, optic canal and cavernous sinus if vision is compromised. A complete neurological assessment is also indicated in these cases, especially if there was loss of consciousness, as the optic neuropathy is just one among other injuries resulting from the traumatic circumstance. Fortunately, most patients with traumatic optic neuropathy enter the medical system through the emergency department prior to optometric or ophthalmologic consultation. In this instance imaging studies will have already been ordered and interpreted by the ER physician.1-11
Ocular treatment presents three options: (1) careful observation, (2) systemic corticosteroid therapy or (3) optic nerve decompression surgery. While a fair number of patients with traumatic optic neuropathy experience some spontaneous improvement of vision, there is great variability in the outcome. Negative prognostic factors include blood in the posterior ethmoid cells, loss of consciousness, older age (i.e. >40) and complete loss of vision at the initial presentation.1,2
In the 1990s, researchers quoting the Second National Acute Spinal Cord Injury Study recommended megadose systemic corticosteroid therapy on all patients with traumatic optic neuropathy within eight hours of injury.12,16,44-59 Those patients remaining unresponsive to the therapy after several days, or those with poorer visual acuity (i.e., finger counting or worse) at initial presentation are considered candidates for optic canal decompression surgery.12,16
Experimental paradigms to promote survival of RGCs and optic nerve regeneration through stimulation via neurotrophic factors (NTF) either directly or indirectly through retinal astrocyte/Müller cell intermediary activation are underway.19 NTFs induce disinhibition of axon growth through regulated intramembranous proteolysis.19 The concomitant release of metalloproteinases (MMPs) and plasminogen activators from RGC axons as well as tissue inhibitors of metalloproteinases from optic nerve glia, repress scarring.19 MMPs also degrade myelin-derived inhibitory messengers along regenerating axon trajectories, encouraging growth. The combination of blocking axon-growth inhibition while stimulating axon growth promoters may be the therapeutic formula for sustained axon regeneration.19
For now, medical and/or surgical intervention remains of questionable value.20 It has been suggested that patients without negative prognostic indicators be effectively managed with careful monitoring.1,17,20 Furthermore, research has shown that there is no significant difference in final visual acuity relating to dose (low vs. high vs. mega) of corticosteroid therapy, there is no significantly different outcome between patients treated with steroids or surgical decompression of the optic canal and in cases presenting more than eight hours after the initial injury, steroids are contraindicated.17,18,20 Patients with traumatic optic neuropathy should, therefore, be managed via the above outlined options on an individual basis, following proper assessment and consultation.17,18,20
Late “traumatic” glaucoma may ensue following any episode of violent ocular injury.
This patient was educated regarding the findings and educated to always protect both eyes from new injuries. They were informed of the limitations of corrective lenses and the need for ongoing monitoring for both late glaucoma as well as choroidal neovascular membrane formation.
Dr. Gurwood thanks Aikaterini Koukas, OD, and James Forde, OD, for their contributions to this case.
Dr. Gurwood is a professor of clinical sciences at The Eye Institute of the Pennsylvania College of Optometry at Salus University. He is a co-chief of Primary Care Suite 3. He is attending medical staff in the department of ophthalmology at Albert Einstein Medical Center, Philadelphia. He has no financial interests to disclose.
|
